Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Programmed cell death is an integral component of C. elegans development. Genetic studies in C. elegans have led to the identification of more than two dozen genes that are important for the specification of which cells should live or die, the activation of the suicide program, and the dismantling and removal of dying cells. Molecular and biochemical studies have revealed the underlying conserved mechanisms that control these three phases of programmed cell death. In particular, an interplay of transcriptional regulatory cascades and networks involving CES-1, CES-2, HLH-1/HLH-2, TRA-1, and other transcriptional regulators is crucial in activating the expression of the key death-inducing gene egl-1 in cells destined to die. A protein interaction cascade involving EGL-1, CED-9, CED-4 and CED-3 results in the activation of the key cell death protease CED-3. The activation of CED-3 initiates the cell disassembly process and nuclear DNA fragmentation, which is mediated by the release of apoptogenic mitochondrial factors (CPS-6 and WAH-1) and which involves multiple endo- and exo-nucleases such as NUC-1 and seven CRN nucleases. The recognition and removal of the dying cell is mediated by two partially redundant signaling pathways involving CED-1, CED-6 and CED-7 in one pathway and CED-2, CED-5, CED-10, CED-12 and PSR-1 in the other pathway. Further studies of programmed cell death in C. elegans will continue to advance our understanding of how programmed cell death is regulated, activated, and executed in multicellular organisms.
Genetic studies of programmed cell death, or apoptosis, in C. elegans led to the identification of key players involved in this important physiological process from C. elegans to humans (Adams, 2003; Danial and Korsmeyer, 2004; Horvitz, 2003). These pioneering studies were made possible by the biology of C. elegans: 1. Unlike in many other animals, programmed cell death is not essential for C. elegans viability, at least under laboratory conditions (Ellis and Horvitz, 1986); 2. cells undergoing programmed cell death in C. elegans change their morphology and refractivity and can be observed in living animals using Differential Interference Contrast microscopy (DIC), also referred to as Nomarski optics (Figure 1; Robertson and Thomson, 1982); 3. programmed cell death that occurs during C. elegans development is determined by the essentially invariant somatic cell lineage of C. elegans; therefore, it is not only known which cells undergo programmed cell death but also when and where they die (Sulston and Horvitz, 1977; Sulston et al., 1983). These unique features made it possible to genetically dissect the process of programmed cell death in C. elegans at single cell resolution. The resulting ground-breaking work was recognized with the Nobel Prize for Medicine in 2002, which was awarded to Sydney Brenner, John E. Sulston, and H. Robert Horvitz for their leading roles in deciphering the C. elegans cell lineage and in defining the genetic pathway of programmed cell death (Brenner, 2003; Horvitz, 2003; Sulston, 2003).
|
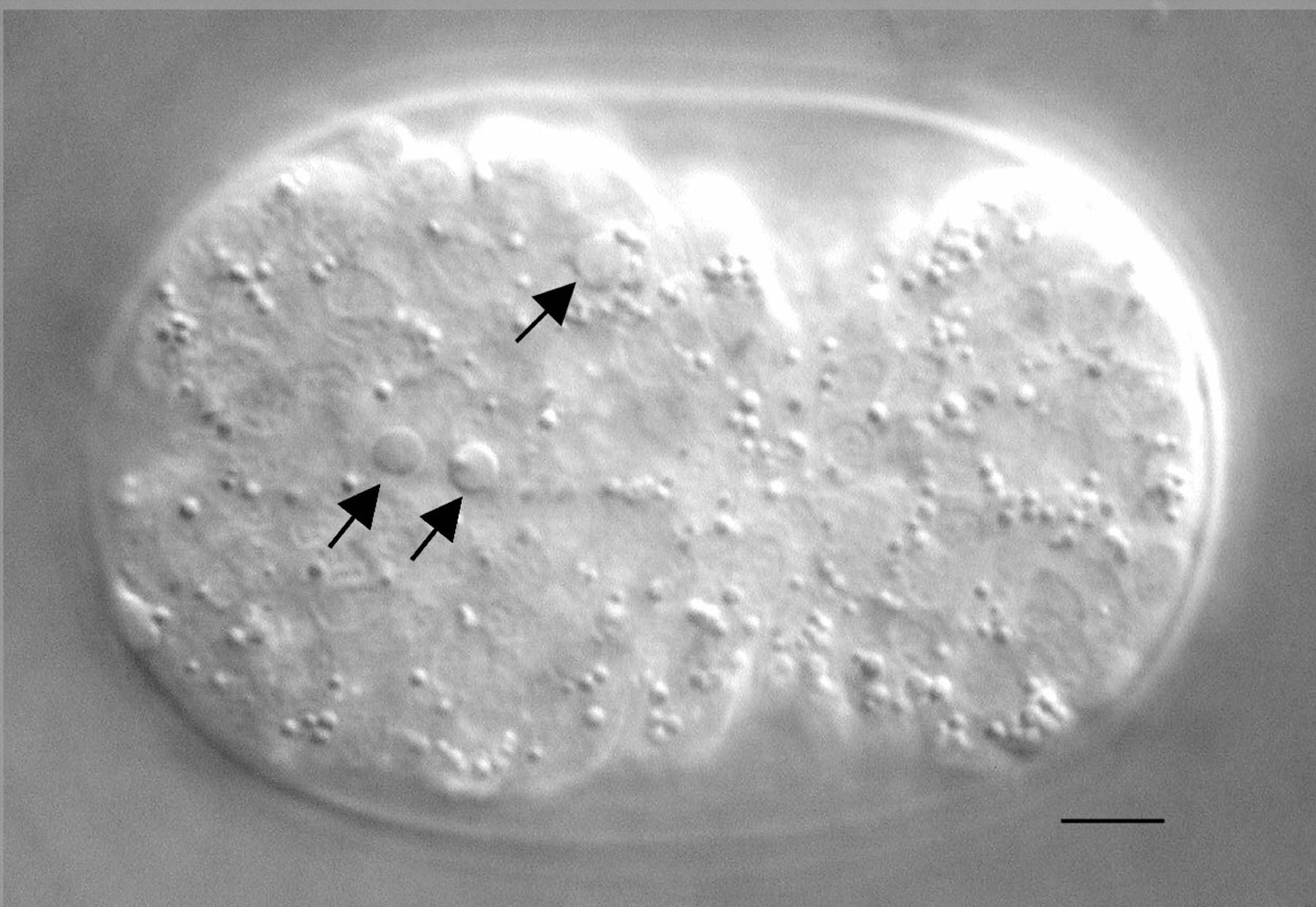
Figure 1. Nomarski image of an embryo with apoptotic cells. Three cells indicated by arrows underwent programmed cell death in a bean/comma stage embryo and exhibit a refractile, raised-button-like appearance. The bar represents 5 μm.
Programmed cell death occurs during two stages of C. elegans life and in two different types of tissues: during embryonic and post-embryonic development of the soma (referred to as "developmental cell death"; Sulston and Horvitz, 1977; Sulston et al., 1983) and in the gonad of adult hermaphrodites (referred to as "germ cell death"; Gumienny et al., 1999; Sulston, 1988; White, 1988). Programmed cell death proceeds in three genetically distinguishable phases: during the "specification phase", a cell is instructed to undergo programmed cell death; in the "killing phase", the apoptotic program is activated in the cell instructed to die; during the "execution phase", cells are dismantled and subsequently engulfed by neighboring cells (Horvitz, 1999; Figure 2). Mutations that lead to a partial block in this final phase, such as mutations in the genes ced-1 and ced-2 (Hedgecock et al., 1983), result in the accumulation of dead cells (referred to as "cell corpses"). Mutations in ced-1 and ced-2 were the first mutations to be identified as affecting programmed cell death and they were instrumental in the subsequent identification of genes involved in apical phases of programmed cell death. In the following, we will review our current understanding of the genes involved in the specification, killing, and execution phases in the case of developmental cell death. Germ cell death is discussed elsewhere in this book (see Germline survival and cell death).
|
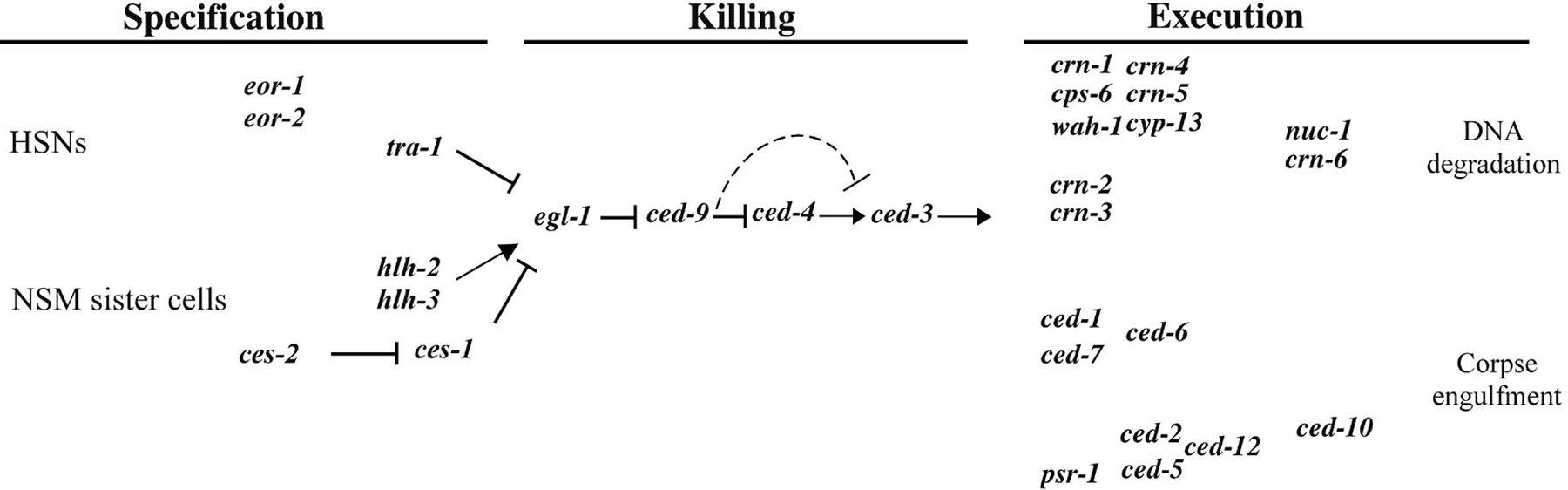
Figure 2. Genetic pathway of programmed cell death in C. elegans. Three phases of programmed cell death, specification, killing, and execution, are indicated. In the "specification" phase, genes involved in regulating the death fates of specific cells (HSNs and NSM sister cells) are shown. In the "execution" phase, two partially redundant pathways mediate the engulfment of cell corpses and the fragmentation of chromosomal DNA. nuc-1 and crn-6 may be involved in the degradation of DNA debris from apoptotic cells.
Three death-promoting genes, egl-1 (egl, egg-laying defective), ced-3 (ced, cell-death abnormal) and ced-4, are required for most, if no all, developmental cell death in C. elegans. Strong loss-of-function (lf) mutations in any of these genes result in the survival of essentially all cells that normally undergo programmed cell death during development (Conradt and Horvitz, 1998; Ellis and Horvitz, 1986). Furthermore, these three genes act within dying cells to promote apoptosis, indicating that cells die by an intrinsic suicide mechanism (Conradt and Horvitz, 1998; Shaham and Horvitz, 1996a; Yuan and Horvitz, 1990). In contrast, the activity of the ced-9 gene protects cells from undergoing programmed cell death during C. elegans development (Hengartner et al., 1992). Loss-of-function mutations in ced-9 cause embryonic lethality, as a consequence of the ectopic deaths of cells that normally live. ced-3, ced-4, egl-1, and ced-9 appear to act in a simple genetic pathway in which egl-1 acts upstream of ced-9 to induce cell death, ced-9 acts upstream of ced-4 to inhibit cell death, and ced-4 acts upstream of ced-3 to kill cells (Conradt and Horvitz, 1998; Hengartner et al., 1992; Shaham and Horvitz, 1996a; Figure 2).
ced-9 encodes a protein similar to the product of the human proto-oncogene bcl-2 (Hengartner and Horvitz, 1994), which plays a similar role in preventing apoptosis in mammals (Adams and Cory, 2001). ced-9 and bcl-2 are members of a gene family that plays important roles in regulating apoptosis in diverse organisms (Adams and Cory, 2001; Reed, 1997). egl-1 encodes a small protein of 91 amino acids with a BH3 (BH3, Bcl-2 homology region 3) motif, which has been found in all pro-apoptotic members of the Bcl-2 gene family and mediates direct binding of these proteins to anti-apoptotic Bcl-2 members (Bouillet and Strasser, 2002; Conradt and Horvitz, 1998). ced-3 encodes the founding member of a family of aspartate-specific cysteine proteases named caspases (Alnemri et al., 1996; Yuan et al., 1993). Like other caspases, CED-3 is synthesized as a proenzyme and is proteolytically activated to generate an active protease containing a p13 and p17 subunit (Alnemri et al., 1996; Xue et al., 1996). CED-3 protease activity appears to be essential for ced-3 to cause programmed cell death in C. elegans (Xue et al., 1996). ced-4 encodes a protein similar to human Apaf-1 (apoptotic protease activating factors), an activator of human caspase-9 (Yuan and Horvitz, 1992; Zou et al., 1997). Both CED-4 and Apaf-1 contain a caspase-recruitment domain (CARD domain) and nucleotide-binding motifs that are critical for the function of these proteins (Seshagiri and Miller, 1997; Zou et al., 1999). By analogy, CED-4 may play a role in activating CED-3 during apoptosis. Interestingly, ced-4 may also produce an alternatively spliced transcript, ced-4L, which encodes a slightly larger protein (CED-4L) with a twenty-four amino acid insertion between its two nucleotide-binding motifs and which might protect against programmed cell death (Shaham and Horvitz, 1996b).
Biochemical and cell biological analyses of EGL-1, CED-9, CED-4, and CED-3 have provided important insights into how these proteins function to regulate the activation of programmed cell death during C. elegans development (Horvitz, 2003). CED-4 has been shown to physically interact with CED-9 in vitro and in cultured cells (Chinnaiyan et al., 1997; Spector et al., 1997; Wu et al., 1997). Furthermore, endogenous CED-9 and CED-4 proteins co-localize at the surface of mitochondria in C. elegans embryos and the mitochondrial localization of CED-4 is dependent on CED-9 (Chen et al., 2000). In addition to CED-9, CED-4 has been shown to interact with CED-3 in vitro and in mammalian cells and it has been proposed that a ternary CED-9/CED-4/CED-3 complex might form in vivo (Chinnaiyan et al., 1997; Yang et al., 1998). However, the subcellular localization of endogenous CED-3 has not been determined. Ectopic egl-1 expression in C. elegans embryos results in the translocation of CED-4 to perinuclear membranes and ectopic programmed cell death (Chen et al., 2000). CED-4 translocation from mitochondria to perinuclear membranes appears to be initiated by the binding of EGL-1 to CED-9, which induces a major conformational change in the CED-9 protein (Yan et al., 2004), resulting in the release of CED-4 from the CED-9/CED-4 complex (Conradt and Horvitz, 1998; del Peso et al., 1998; Parrish et al., 2000). A gain-of-function mutation in ced-9 (n1950), which results in the substitution of glycine 169 with glutamate and which blocks most, if not all programmed cell death during development, impairs the binding of EGL-1 to CED-9 and EGL-1-induced release of CED-4 (Parrish et al., 2000; Yan et al., 2004). EGL-1-induced CED-4 translocation is thought to be important for the activation of CED-3 and hence programmed cell death (Chen et al., 2000). CED-4 translocation may promote CED-4 self-oligomerization, which is thought to bring CED-3 proenzymes to close proximity for self-activation (Yang et al., 1998). The only C. elegans substrate of CED-3 known so far is CED-9 (Xue and Horvitz, 1997). CED-9 may not only inhibit cell death activation by sequestering CED-4 to mitochondria but also by acting as a competitive inhibitor of CED-3 (Xue and Horvitz, 1997). The mechanism by which CED-3 is activated appears to differ somewhat from the mechanisms that activate mammalian caspases, which involve either release of Cytochrome c from mitochondria and assembly of an oligomerized Apaf-1/caspase-9 apoptosome (caspase-9 activation), the formation of caspase-8 trimers induced by activation of death receptors (caspases-8 activation), or direct proteolytic activation of downstream executor caspases (such as caspase-3 and caspase-6) by upstream initiator caspases (such as caspase-8 and caspase-9; Budihardjo et al., 1999; Jiang and Wang, 2004; Liu et al., 1996). The C. elegans genome has three additional caspase-like genes but it is currently unclear if any of these genes is involved in programmed cell death (Shaham, 1998).
In addition to egl-1, ced-3, ced-4 and ced-9, several other genes have been implicated in the activation of the apoptotic program during C. elegans development, including the dad-1 gene (dad, defender against apopototic death; Sugimoto et al., 1995), which encodes a protein similar to the mammalian DAD1 protein (Nakashima et al., 1993), and the icd-1 gene (icd-1, inhibitor of cell death; Bloss et al., 2003; Sugimoto et al., 1995), which encodes a protein similar to the beta-subunit of the nascent polypeptide-associated complex (betaNAC). How dad-1 and icd-1 might interact with the core killing machinery is currently unclear.
|
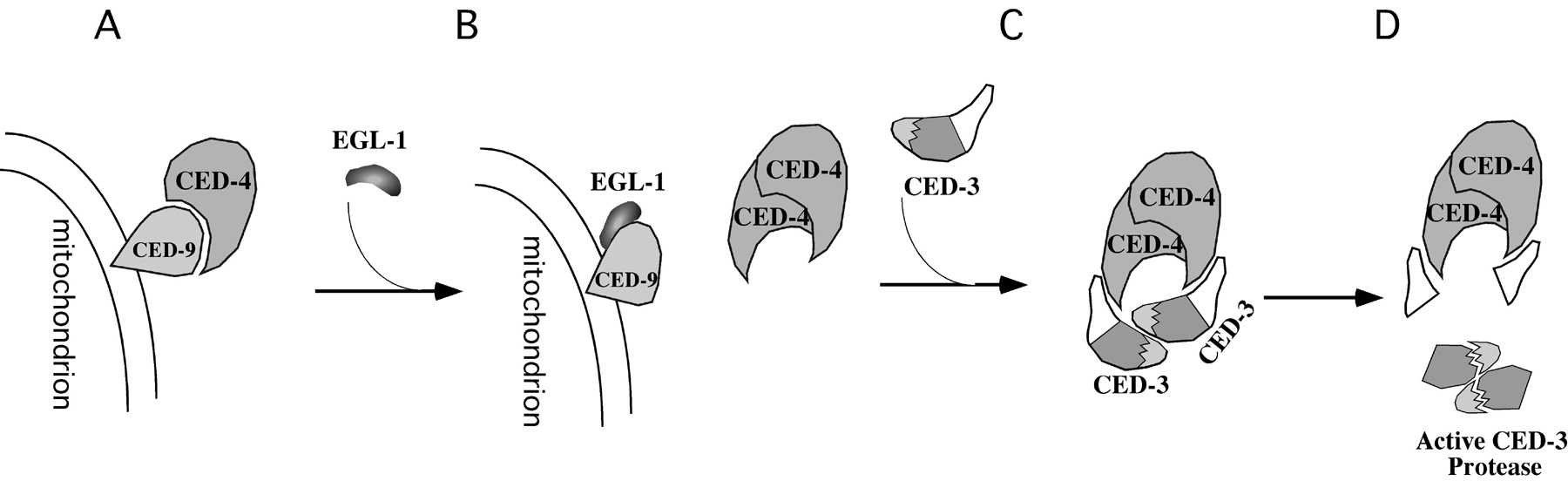
Figure 3. Biochemical model for the activation of programmed cell death. (A) In living cells, CED-4 is tethered to the surface of mitochondria through binding to CED-9. (B) In cells that are doomed to die, the death initiator EGL-1 binds to CED-9, causes a major CED-9 conformational change, and triggers the disassociation of CED-4 from CED-9. (C) Released CED-4 proteins translocate to perinuclear membranes and undergo oligomerization, which brings two CED-3 proenzymes to close proximity. (D) CED-3 proenzymes undergo autoproteolytic activation.
Out of the 1090 cells generated during the development of the soma of a C. elegans hermaphrodite, 131 undergo programmed cell death (113 of these cells die during embryonic and 18 during post-embryonic development; Sulston and Horvitz, 1977; Sulston et al., 1983). If prevented from undergoing programmed cell death, the majority of these cells adopt a neuronal fate, indicating that they are of neuronal origin (Ellis and Horvitz, 1986; Ellis and Horvitz, 1991; White et al., 1991). At least some of these "undead" neurons appear to be fully functional (Avery and Horvitz, 1987).
How does a cell know whether to live or die? In higher organisms, cells are generally instructed to die by neighboring cells or by extracellular cues. In contrast, most of the 131 cell deaths observed during C. elegans development appear to occur in a cell-autonomous manner; i.e. cells appear to "know" at the time of their birth whether their "fate" is to live or die (Sulston and White, 1980; Evidence for cell non-autonomous induction of cell death exists however in the germ line of C. elegans hermaphrodites; see Germline survival and cell death). The observations that cell fate altering mutations, such as loss-of-function mutations of unc-86 (unc, uncoordinated) or pag-3 (pag, pattern of reporter gene expression abnormal), can affect the essentially invariant pattern of programmed cell death suggest that programmed cell death can be regarded as a cell fate (Cameron et al., 2002; Chalfie et al., 1981; Finney et al., 1988; Sulston and Horvitz, 1981). The current model for cell death specification is that in the 959 cells destined to survive, EGL-1 activity is low or absent and that in the 131 cells destined to die, EGL-1 activity is high. High EGL-1 activity inhibits CED-9 activity, resulting in the activation of CED-4 and CED-3 and the commitment of a cell to the cell death fate (Horvitz, 2003).
EGL-1 activity appears to be regulated at the level of transcription. The egl-1 gene is expressed predominantly in cells destined to die (Conradt and Horvitz, 1999; Thellmann et al., 2003). Furthermore, mutations in cis-acting regions of the egl-1 locus cause changes in the essentially invariant pattern of developmental cell death (Conradt and Horvitz, 1999). Depending on the particular cells, different transcriptional regulators appear to control egl-1 expression. For instance, the Zn finger DNA-binding protein and transcriptional repressor TRA-1 (TRA, transformer; Hodgkin, 1987; Zarkower and Hodgkin, 1992) represses egl-1 expression specifically in the two hermaphrodite-specific neurons (HSNs; Conradt and Horvitz, 1999; Figure 2). Furthermore, the transcriptional activator HLH-2/HLH-3 (HLH, helix-loop-helix transcription factor family), a heterodimer composed of the basic helix-loop-helix (bHLH) proteins HLH-2 and HLH-3 (Krause et al., 1997), is at least partially required for the egl-1-dependent death of the NSM sister cells (NSM, neuro-secretory motorneuron; Thellmann et al., 2003). Finally, the is- or overexpression of the Snail-like Zn finger DNA-binding protein and transcriptional repressor CES-1 (CES, cell-death selection abnormal) in the NSM sister cells inappropriately blocks egl-1 expression thereby preventing the death of these cells (Metzstein and Horvitz, 1999; Thellmann et al., 2003). ces-1 expression in the NSM sister cells might normally be repressed by the basic leucine zipper (bZIP) DNA-binding protein CES-2 (Metzstein et al., 1996). These observations suggest that the pattern of egl-1 expression is the result of the interplay of cascades or networks of positive and negative regulators of transcription. HLH-2/HLH-3, CES-1, and TRA-1 proteins can bind to cis-acting regions of the egl-1 promoter in vitro that have been shown to be important for controlling the death of the NSM sister cells (HLH-2/HLH-3, CES-1) or the HSNs (TRA-1) in vivo and that are conserved in a related Caenorhabditis species, C. briggsae (Conradt and Horvitz, 1999; Thellmann et al., 2003). Hence, HLH-2/HLH-3, CES-1, and TRA-1 might be direct regulators of egl-1 transcription.
At least some of the regulators of egl-1 expression have additional functions during development. For example, TRA-1 is the terminal, global regulator of somatic sexual fate required for female development (Hodgkin, 1987; Zarkower and Hodgkin, 1992) and HLH-2 is essential for viability (Krause et al., 1997). Some of the cascades or networks that control egl-1 expression during development appear to be conserved: CES-1-like members of the Snail family of DNA-binding proteins, such as Snail and SLUG, can confer resistance to programmed cell death in mammals and in chick (Inoue et al., 2002; Inukai et al., 1999; Perez-Losada et al., 2003; Vega et al., 2004), and the CES-2-like proto-oncogene HLF (HLF, hepatic leukemia factor) has been implicated in the regulation of SLUG in mammals (Inaba et al., 1996; Inukai et al., 1999). However, egl-1 might not be the only gene that needs to be regulated at the transcriptional level for proper cell death specification. The proteins EOR-1 and EOR-2 (EOR, enhancer of Raf), which have been proposed to act as regulators of transcription (Howard and Sundaram, 2002), are required for the death of the HSNs in males, however, they are not required for the activation of egl-1 transcription in these cells (Hoeppner et al., 2004).
Once the apoptotic program is activated, it initiates the cell disassembly process, which includes nuclear DNA fragmentation, cytoplasm shrinkage, and exposure of "eat-me" signal(s) on the cell surface to induce phagocytosis by neighboring cells (Steller, 1995).
The fragmentation of chromosomal DNA is a hallmark of apoptosis and may facilitate apoptosis by terminating DNA replication and gene transcription (Arends et al., 1990). DNA fragmentation during C. elegans apoptosis has been studied with the aid of various DNA-staining techniques, including DAPI or Feulgen staining (Sulston, 1976) or TUNEL staining (Gavrieli et al., 1992; Wu et al., 2000).
So far ten genes have been identified to be involved in nuclear DNA degradation during apoptosis (Parrish et al., 2001; Parrish and Xue, 2003; Sulston, 1976; Wang et al., 2002; Wu et al., 2000). These include nuc-1 (nuc, nuclease defective), cps-6 (cps, CED-3 protease suppressors), wah-1 (wah, worm AIF homologue), crn-1 to crn-6 (crn, cell death related nucleases), and cyp-13 (cyp, cyclophilins). Loss or reduction of activity in any of these genes results in the accumulation of TUNEL-positive cells in C. elegans embryos, suggesting that these genes are important for resolving TUNEL-reactive DNA breaks generated during apoptosis (Parrish et al., 2001; Parrish and Xue, 2003; Sulston, 1976; Wang et al., 2002; Wu et al., 2000). In addition, reduction of activity in most of these genes (with the exception of nuc-1 and crn-6) causes delayed appearance of embryonic cell corpses during development and reduced cell deaths in sensitized genetic backgrounds, suggesting that nuclear DNA degradation is important for normal progression of the apoptotic process and can even promote cell killing. Genetic and phenotypic analyses indicate that these genes act in multiple pathways and at different stages to promote DNA degradation and apoptosis, with cps-6, wah-1, crn-1, crn-4, crn-5 and cpy-13 acting in one pathway and crn-2 and crn-3 in the other (Parrish and Xue, 2003; Wang et al., 2002). Defects in both DNA degradation pathways not only cause a more severe defect in nuclear DNA degradation but also a synthetic defect in cell corpse engulfment, suggesting that the DNA degradation process may affect cell corpse removal (Parrish and Xue, 2003). In addition to its cell death function, nuc-1 is involved in the degradation of DNA derived from ingested bacteria in the intestinal lumens (Sulston, 1976; Wu et al., 2000).
Both cps-6 and wah-1 encode mitochondrial proteins, which are similar to human mitochondrial endonuclease G (EndoG) and apoptosis-inducing factor (AIF), respectively (Parrish et al., 2001; Wang et al., 2002). Ectopic egl-1 expression induces WAH-1 translocation from mitochondria to nuclei in a CED-3 dependent manner, suggesting that the role of mitochondria in regulating apoptosis is conserved. The WAH-1 protein can physically associate with CPS-6 and enhance the endonuclease activity of CPS-6 (Wang et al., 2002). Furthermore, CPS-6, CRN-1, CRN-4, CRN-5 and CYP-13, which are either endonucleases or exonucleases, appear to interact and cooperate with one another, possibly in a large DNA degradation complex named degradeosome (Parrish and Xue, 2003), to promote stepwise DNA fragmentation, starting from generating DNA nicks, gaps, to double-stranded DNA breaks (Parrish et al., 2003). Both nuc-1 and crn-6 encode type II acidic DNases and do not seem to affect either the activation or progression of cell death or the engulfment of cell corpses (Hedgecock et al., 1983; Parrish et al., 2001; Parrish and Xue, 2003; Wu et al., 2000). These two nucleases may act at later stages of apoptotic DNA degradation, possibly in the lysosomal compartments of the engulfing cell to promote degradation of engulfed apoptotic cells.
When a cell undergoes apoptosis, "eat-me" signals are rapidly exposed on the surface of the dying cell, which are recognized by engulfing cells (Fadok et al., 2001). The engulfment signal is transduced to the cellular machinery in the phagocyte to trigger the phagocytic process (Hedgecock et al., 1983; Sulston and Horvitz, 1977). Genetic analyses have identified eight genes, ced-1, ced-2, ced-5, ced-6, ced-7, ced-10, ced-12, and psr-1 (psr, phosphatidylserine receptor homologue), which appear to function in two pathways to promote the cell-corpse engulfment process, with ced-1, ced-6 and ced-7 acting in one pathway and ced-2, ced-5, ced-10, ced-12 and psr-1 in the other pathway (Chung et al., 2000; Ellis et al., 1991; Gumienny et al., 2001; Wang et al., 2003; Wu et al., 2001; Zhou et al., 2001). Most of these genes act in engulfing cells to promote corpse removal, with the exception of ced-7 whose activity is required in both the dying cell and the engulfing cells (Reddien and Horvitz, 2004; Wu and Horvitz, 1998a).
ced-1, ced-6 and ced-7 appear to encode components of a signaling pathway involved in cell-corpse recognition. CED-1 is similar to the human scavenger receptor SREC and may function as a corpse-recognizing phagocytic receptor since CED-1 protein is found to cluster around cell corpses (Zhou et al., 2001). CED-7 is similar to ABC (ATP-binding cassette) transporters and may play a role in promoting or mediating cell-corpse recognition by CED-1 as CED-1 receptors fail to cluster around dying cells in mutants defective in the ced-7 gene (Wu and Horvitz, 1998a; Zhou et al., 2001). The CED-6 protein contains a PTB (phosphotyrosine-binding) domain (Liu and Hengartner, 1999), which directly binds to the intracellular domain of CED-1 (Su et al., 2002). CED-6 may act as a signaling adaptor downstream of CED-1 and CED-7 (Figure 4).
|
Figure 4. Molecular model for the cell corpse engulfment process. The engulfment process is mediated by two partially redundant pathways. In the CED-1/CED-6/CED-7 pathway, CED-1 and CED-7 act on the surface of the engulfing cell to mediate recognition of an unknown engulfment signal(s) on the surface of the dying cell (green diamonds) and to transduce the signal through CED-6 to activate the phagocytic machinery of the engulfing cell. CED-7 also acts in dying cells. In the CED-2, CED-5, CED-10 and CED-12 pathway, PSR-1 may act in the engulfing cell to mediate the recognition of PS (red circles) externalized by the dying cell and to transduce the signal through the CED-2/CED-5/CED-12 ternary complex to activate CED-10. There are likely other engulfment receptors that act in the CED-2, CED-5, CED-10 and CED-12 pathway.
In addition to mediating corpse engulfment, ced-2, ced-5, ced-10, and ced-12 control the migration of the gonadal distal tip cells (DTC) and the CAN cells (Gumienny et al., 2001; Reddien and Horvitz, 2000; Wu and Horvitz, 1998a; Wu et al., 2001; Zhou et al., 2001), and the development of D-type motorneurons and amphid sensory neurons (Lundquist et al., 2001; Wu et al., 2002). ced-2, ced-5, ced-10, and ced-12 encode conserved components of the Rac GTPase signaling pathway involved in regulating actin cytoskeleton rearrangement essential for cell migration and corpse engulfment. CED-2 is a CrkII-like adaptor with one SH2 and two SH3 domains (Reddien and Horvitz, 2000). CED-5 is similar to human DOCK180, which physically interacts with human CrkII (Wu and Horvitz, 1998a). CED-10 is a C. elegans homologue of mammalian Rac GTPase (Reddien and Horvitz, 2000), which controls cytoskeletal dynamics and cell shape changes. CED-12 contains a potential PH (pleckstrin-homology) domain and an SH3-binding motif (Gumienny et al., 2001; Wu et al., 2001; Zhou et al., 2001). Genetic analyses indicate that ced-2, ced-5 and ced-12 appear to function at the same step but upstream of ced-10 in the engulfment process. Biochemical analyses suggest that CED-2, CED-5 and CED-12 form a ternary complex to activate CED-10 GTPase activity (Brugnera et al., 2002; Chung et al., 2000; Ellis et al., 1991; Gumienny et al., 2001; Lu et al., 2004; Wu et al., 2001; Zhou et al., 2001). The phosphatidylserine receptor-like protein PSR-1 specifically recognizes apoptotic cells with externalized phosphatidylserine (PS), an engulfment-inducing signal (Fadok et al., 2001), and may trigger the formation of the CED-2/CED-5/CED-12 ternary complex and the subsequent activation of CED-10 GTPase by binding to CED-5 and CED-12. Since the psr-1 mutant displays a significantly weaker engulfment defect than any of the ced-2, ced-5, ced-10 mutants, other "corpse-recognizing" receptor(s) must also act in this pathway (Wang et al., 2003; Figure 4). In addition to CED-10, two other Rac-like GTPases, MIG-2 and RAC-2, also contribute to cell corpse phagocytosis (Lundquist et al., 2001).
Phagocytosis not only removes cell corpses generated by programmed cell death, but also may actively promote apoptosis (Hoeppner et al., 2001; Reddien et al., 2001). Engulfment genes appear to act in engulfing cells to promote apoptosis (Reddien et al., 2001). Furthermore, these two engulfment pathways are also important for the removal of necrotic cell corpses, suggesting that similar mechanisms are used to recognize and remove apoptotic and necrotic corpses (Chung et al., 2000). Several other genes have also been implicated in cell death execution, including the ced-8 gene which appears to control the timing of cell death and encodes a protein similar to human XK, a putative membrane transport protein (Stanfield and Horvitz, 2000).
The genetic and molecular characterization of genes, which, when mutated or inactivated by RNAi, affect developmental cell death in C. elegans, has revealed some of the molecular mechanisms involved in the specification, killing or execution phase of programmed cell death. However, much remains to be learnt about programmed cell death. For example, while we know that the transcriptional activation of the egl-1 gene specifies whether a cell will live or die, very little is known about what regulates egl-1 expression. Furthermore, while we know that the CED-3 caspase is essential for cell killing and required for DNA fragmentation and engulfment, it is currently not known how CED-3 is activated and what CED-3 substrates are. Finally, while more is known about the molecular components that act in engulfing cells to mediate cell corpse engulfment, little is known about what acts in the dying cell to trigger the phagocytic event and how engulfing cells promote killing in a cell non-autonomous manner. By answering these and other remaining questions, studies of developmental cell death in C. elegans will continue to contribute in a major way to our current knowledge of programmed cell death.
We would like to thank Yi-chun Wu for contributing some of the figures and for writing a portion of this chapter. Research in Ding Xue's laboratory is supported by a Searle Scholar award, a Burroughs Wellcome Fund Career Award, and grants from NIH and Department of Defense. Research in Barbara Conradt's laboratory is supported by the Howard Hughes Medical Institute (HHMI award to Dartmouth Medical School under the Biomedical Research Support Program for Medical Schools) and NIH.
Adams, J.M. (2003). Ways of dying: multiple pathways to apoptosis. Genes Dev. 17, 2481–2495. Abstract Article
Adams, J.M., and Cory, S. (2001). Life-or-death decisions by the Bcl-2 protein family. Trends Biochem Sci. 26, 61–66. Abstract Article
Alnemri, E.S., Livingston, D.J., Nicholson, D.W., Salvesen, G., Thornberry, N.A., Wong, W.W., and Yuan, J. (1996). Human ICE/CED-3 protease nomenclature. Cell 87, 171. Abstract Article
Arends, M.J., Morris, R.G., and Wyllie, A.H. (1990). Apoptosis. The role of the endonuclease. Am. J. Pathol. 136, 593–608. Abstract
Avery, L., and Horvitz, H.R. (1987). A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell 51, 1071–1078. Abstract Article
Bloss, T.A., Witze, E.S., and Rothman, J.H. (2003). Suppression of CED-3-independent apoptosis by mitochondrial betaNAC in Caenorhabditis elegans. Nature 424, 1066–1071. Abstract Article
Bouillet, P., and Strasser, A. (2002). BH3-only proteins—evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J. Cell Sci. 115, 1567–1574. Abstract
Brenner, S. (2003). Nobel lecture. Nature's gift to science. Biosci. Rep. 23, 225–237. Abstract Article
Brugnera, E., Haney, L., Grimsley, C., Lu, M., Walk, S.F., Tosello-Trampont, A.C., Macara, I.G., Madhani, H., Fink, G.R., and Ravichandran, K.S. (2002). Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat. Cell Biol. 4, 574–582. Abstract Article
Budihardjo, I., Oliver, H., Lutter, M., Luo, X., and Wang, X. (1999). Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290. Abstract Article
Cameron, S., Clark, S.G., McDermott, J.B., Aamodt, E., and Horvitz, H.R. (2002). PAG-3, a Zn-finger transcription factor, determines neuroblast fate in C. elegans. Development 129, 1763–1774. Abstract
Chalfie, M., Horvitz, H.R., and Sulston, J.E. (1981). Mutations that lead to reiterations in the cell lineages of C. elegans. Cell 24, 59–69. Abstract Article
Chen, F., Hersh, B.M., Conradt, B., Zhou, Z., Riemer, D., Gruenbaum, Y., and Horvitz, H.R. (2000). Translocation of C. elegans CED-4 to nuclear membranes during programmed cell death. Science 287, 1485–1489. Abstract Article
Chinnaiyan, A.M., O'Rourke, K., Lane, B.R., and Dixit, V.M. (1997). Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science 275, 1122–1126. Abstract Article
Chung, S., Gumienny, T.L., Hengartner, M.O., and Driscoll, M. (2000). A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat. Cell Biol. 2, 931–937. Abstract Article
Conradt, B., and Horvitz, H.R. (1998). The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93, 519–529. Abstract Article
Conradt, B., and Horvitz, H.R. (1999). The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell 98, 317–327. Abstract Article
Danial, N.N., and Korsmeyer, S.J. (2004). Cell death: critical control points. Cell 116, 205–219. Abstract Article
del Peso, L., Gonzalez, V.M., and Nunez, G. (1998). Caenorhabditis elegans EGL-1 disrupts the interaction of CED-9 with CED-4 and promotes CED-3 activation. J. Biol. Chem. 273, 33495–33500. Abstract Article
Ellis, H.M., and Horvitz, H.R. (1986). Genetic control of programmed cell death in the nematode C. elegans. Cell 44, 817–829. Abstract Article
Ellis, R.E., and Horvitz, H.R. (1991). Two C. elegans genes control the programmed deaths of specific cells in the pharynx. Development 112, 591–603. Abstract
Ellis, R.E., Jacobson, D.M., and Horvitz, H.R. (1991). Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics 129, 79–94. Abstract
Fadok, V.A., Xue, D., and Henson, P. (2001). If phosphatidylserine is the death knell, a new phosphatidylserine-specific receptor is the bellringer. Cell Death Differ. 8, 582–587. Abstract Article
Finney, M., Ruvkun, G., and Horvitz, H.R. (1988). The C. elegans cell lineage and differentiation gene unc-86 encodes a protein with a homeodomain and extended similarity to transcription factors. Cell 55, 757–769. Abstract Article
Gavrieli, Y., Sherman, Y., and Ben-Sasson, S.A. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493–501. Abstract Article
Gumienny, T.L., Brugnera, E., Tosello-Trampont, A.C., Kinchen, J.M., Haney, L.B., Nishiwaki, K., Walk, S.F., Nemergut, M.E., Macara, I.G., Francis, R., et al. (2001). CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107, 27–41. Abstract Article
Hedgecock, E.M., Sulston, J.E., and Thomson, J.N. (1983). Mutations affecting programmed cell deaths in the nematode Caenorhabditis elegans. Science 220, 1277–1279. Abstract
Hengartner, M.O., Ellis, R.E., and Horvitz, H.R. (1992). Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature 356, 494–499. Abstract Article
Hengartner, M.O., and Horvitz, H.R. (1994). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665–676. Abstract Article
Hodgkin, J. (1987). A genetic analysis of the sex-determining gene, tra-1, in the nematode Caenorhabditis elegans. Genes Dev. 1, 731–745. Abstract
Hoeppner, D.J., Hengartner, M.O., and Schnabel, R. (2001). Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature 412, 202–206. Abstract Article
Hoeppner, D.J., Spector, M.S., Ratliff, T.M., Kinchen, J.M., Granat, S., Lin, S.C., Bhusri, S.S., Conradt, B., Herman, M.A., and Hengartner, M.O. (2004). eor-1 and eor-2 are required for cell-specific apoptotic death in C. elegans. Dev. Biol. 274, 125–138. Abstract Article
Horvitz, H.R. (1999). Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 59, 1701s–1706s. Abstract
Horvitz, H.R. (2003). Worms, life, and death (Nobel lecture). Chembiochem 4, 697–711. Abstract Article
Howard, R.M., and Sundaram, M.V. (2002). C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 mediator component. Genes Dev. 16, 1815–1827. Abstract Article
Inaba, T., Inukai, T., Yoshihara, T., Seyschab, H., Ashmun, R.A., Canman, C.E., Laken, S.J., Kastan, M.B., and Look, A.T. (1996). Reversal of apoptosis by the leukaemia-associated E2A-HLF chimaeric transcription factor. Nature 382, 541–544. Abstract Article
Inoue, A., Seidel, M.G., Wu, W., Kamizono, S., Ferrando, A.A., Bronson, R.T., Iwasaki, H., Akashi, K., Morimoto, A., Hitzler, J.K., et al. (2002). Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2, 279–288. Abstract Article
Jiang, X., and Wang, X. (2004). Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106. Abstract Article
Krause, M., Park, M., Zhang, J.M., Yuan, J., Harfe, B., Xu, S.Q., Greenwald, I., Cole, M., Paterson, B., and Fire, A. (1997). A C. elegans E/Daughterless bHLH protein marks neuronal but not striated muscle development. Development 124, 2179–2189. Abstract
Liu, Q.A., and Hengartner, M.O. (1999). Human CED-6 encodes a functional homologue of the Caenorhabditis elegans engulfment protein CED-6. Curr. Biol. 9, 1347–1350. Abstract Article
Liu, X., Kim, C.N., Yang, J., Jemmerson, R., and Wang, X. (1996). Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157. Abstract Article
Lu, M., Kinchen, J.M., Rossman, K.L., Grimsley, C., de Bakker, C., Brugnera, E., Tosello-Trampont, A.C., Haney, L.B., Klingele, D., Sondek, J., et al. (2004). PH domain of ELMO functions in trans to regulate Rac activation via Dock180. Nat. Struct. Mol. Biol. 11, 756–762. Abstract Article
Lundquist, E.A., Reddien, P.W., Hartwieg, E., Horvitz, H.R., and Bargmann, C.I. (2001). Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development 128, 4475–4488. Abstract
Metzstein, M.M., Hengartner, M.O., Tsung, N., Ellis, R.E., and Horvitz, H.R. (1996). Transcriptional regulator of programmed cell death encoded by Caenorhabditis elegans gene ces-2. Nature 382, 545–547. Abstract Article
Metzstein, M.M., and Horvitz, H.R. (1999). The C. elegans cell death specification gene ces-1 encodes a snail family zinc finger protein. Mol. Cell 4, 309–319. Abstract Article
Nakashima, T., Sekiguchi, T., Kuraoka, A., Fukushima, K., Shibata, Y., Komiyama, S., and Nishimoto, T. (1993). Molecular cloning of a human cDNA encoding a novel protein, DAD1, whose defect causes apoptotic cell death in hamster BHK21 cells. Mol. Cell Biol. 13, 6367–6374. Abstract
Parrish, J., Li, L., Klotz, K., Ledwich, D., Wang, X., and Xue, D. (2001). Mitochondrial endonuclease G is important for apoptosis in C. elegans. Nature 412, 90–94. Abstract Article
Parrish, J., Metters, H., Chen, L., and Xue, D. (2000). Demonstration of the in vivo interaction of key cell death regulators by structure-based design of second-site suppressors. Proc. Natl. Acad. Sci. USA 97, 11916–11921. Abstract Article
Parrish, J.Z., and Xue, D. (2003). Functional genomic analysis of apoptotic DNA degradation in C. elegans. Mol. Cell 11, 987–996. Abstract Article
Parrish, J.Z., Yang, C., Shen, B., and Xue, D. (2003). CRN-1, a Caenorhabditis elegans FEN-1 homologue, cooperates with CPS-6/EndoG to promote apoptotic DNA degradation. EMBO J. 22, 3451–3460. Abstract Article
Perez-Losada, J., Sanchez-Martin, M., Perez-Caro, M., Perez-Mancera, P.A., and Sanchez-Garcia, I. (2003). The radioresistance biological function of the SCF/kit signaling pathway is mediated by the zinc-finger transcription factor Slug. Oncogene 22, 4205–4211. Abstract Article
Reddien, P.W., Cameron, S., and Horvitz, H.R. (2001). Phagocytosis promotes programmed cell death in C. elegans. Nature 412, 198–202. Abstract Article
Reddien, P.W., and Horvitz, H.R. (2000). CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat. Cell Biol. 2, 131–136. Abstract Article
Reddien, P.W., and Horvitz, H.R. (2004). The engulfment process of programmed cell death in Caenorhabditis elegans. Annu. Rev. Cell Dev. Biol. 20, 193–221. Abstract
Reed, J.C. (1997). Double identity for proteins of the Bcl-2 family. Nature 387, 773–776. Article
Robertson, A.G., and Thomson, J.N. (1982). Morphology of programmed cell death in the ventral nerve cord of Caenorhabiditis elegans larvae. J. Embryol. Exp. Morphol. 67, 89–100. Abstract Article
Seshagiri, S., and Miller, L.K. (1997). Caenorhabditis elegans CED-4 stimulates CED-3 processing and CED-3-induced apoptosis. Curr. Biol. 7, 455–460. Abstract Article
Shaham, S. (1998). Identification of multiple Caenorhabditis elegans caspases and their potential roles in proteolytic cascades. J. Biol. Chem. 273, 35109–35117. Abstract Article
Shaham, S., and Horvitz, H.R. (1996a). Developing Caenorhabditis elegans neurons may contain both cell-death protective and killer activities. Genes Dev. 10, 578–591. Abstract
Shaham, S., and Horvitz, H.R. (1996b). An alternatively spliced C. elegans ced-4 RNA encodes a novel cell death inhibitor. Cell 86, 201–208. Abstract Article
Spector, M.S., Desnoyers, S., Hoeppner, D.J., and Hengartner, M.O. (1997). Interaction between the C. elegans cell-death regulators CED-9 and CED-4. Nature 385, 653–656. Abstract Article
Stanfield, G.M., and Horvitz, H.R. (2000). The ced-8 gene controls the timing of programmed cell deaths in C. elegans. Mol. Cell 5, 423–433. Article
Steller, H. (1995). Mechanisms and genes of cellular suicide. Science 267, 1445–1449. Abstract
Su, H.P., Nakada-Tsukui, K., Tosello-Trampont, A.C., Li, Y., Bu, G., Henson, P.M., and Ravichandran, K.S. (2002). Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J. Biol. Chem. 277, 11772–11779. Abstract Article
Sugimoto, A., Hozak, R.R., Nakashima, T., Nishimoto, T., and Rothman, J.H. (1995). dad-1, an endogenous programmed cell death suppressor in Caenorhabditis elegans and vertebrates. EMBO J. 14, 4434–4441. Abstract
Sulston, J.E. (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans R. Soc Lond B. Biol. Sci. 275, 287–297. Abstract
Sulston, J.E. (1988). Cell lineage. In The Nematode Caenorhabditis elegans. (Cold Spring Harbor Laboratory Press), pp. 123–156.
Sulston, J.E. (2003). Caenorhabditis elegans: the cell lineage and beyond (Nobel lecture). Chembiochem. 4, 688–696. Abstract Article
Sulston, J.E., and Horvitz, H.R. (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 56, 110–156. Abstract Article
Sulston, J.E., and Horvitz, H.R. (1981). Abnormal cell lineages in mutants of the nematode Caenorhabditis elegans. Dev. Biol. 82, 41–55. Abstract Article
Sulston, J.E., Schierenberg, E., White, J.G., and Thomson, J.N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119. Abstract Article
Sulston, J.E., and White, J.G. (1980). Regulation and cell autonomy during postembryonic development of Caenorhabditis elegans. Dev. Biol. 78, 577–597. Abstract Article
Thellmann, M., Hatzold, J., and Conradt, B. (2003). The snail-like CES-1 protein of C. elegans can block the expression of the BH3-only cell-death activator gene egl-1 by antagonizing the function of bHLH proteins. Development 130, 4057–4071. Abstract Article
Vega, S., Morales, A.V., Ocana, O.H., Valdes, F., Fabregat, I., and Nieto, M.A. (2004). Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 18, 1131–1143. Abstract Article
Wang, X., Wu, Y.C., Fadok, V.A., Lee, M.C., Gengyo-Ando, K., Cheng, L.C., Ledwich, D., Hsu, P.K., Chen, J.Y., Chou, B.K., et al. (2003). Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science 302, 1563–1566. Abstract Article
Wang, X., Yang, C., Chai, J., Shi, Y., and Xue, D. (2002). Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science 298, 1587–1592. Abstract Article
White, J. (1988). The anatomy. In The Nematode Caenorhabditis elegans. (Cold Spring Harbor Laboratory Press), pp. 81–122.
White, J.G., Southgate, E., and Thomson, J.N. (1991). On the nature of undead cells in the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. 263–271.
Wu, D., Wallen, H.D., and Nunez, G. (1997). Interaction and regulation of subcellular localization of CED-4 by CED-9. Science 275, 1126–1129. Abstract Article
Wu, Y.C., Cheng, T.W., Lee, M.C., and Weng, N.Y. (2002). Distinct rac activation pathways control Caenorhabditis elegans cell migration and axon outgrowth. Dev. Biol. 250, 145–155. Abstract Article
Wu, Y.C., and Horvitz, H.R. (1998a). The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell 93, 951–960. Abstract Article
Wu, Y.C., and Horvitz, H.R. (1998b). C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature 392, 501–504. Article
Wu, Y.C., Stanfield, G.M., and Horvitz, H.R. (2000). NUC-1, a Caenorhabditis elegans DNase II homolog, functions in an intermediate step of DNA degradation during apoptosis. Genes Dev. 14, 536–548. Abstract
Wu, Y.C., Tsai, M.C., Cheng, L.C., Chou, C.J., and Weng, N.Y. (2001). C. elegans CED-12 acts in the conserved crkII/DOCK180/Rac pathway to control cell migration and cell corpse engulfment. Dev. Cell 1, 491–502. Abstract Article
Xue, D., and Horvitz, H.R. (1997). Caenorhabditis elegans CED-9 protein is a bifunctional cell-death inhibitor. Nature 390, 305–308. Abstract Article
Xue, D., Shaham, S., and Horvitz, H.R. (1996). The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 10, 1073–1083. Abstract
Yan, N., Gu, L., Kokel, D., Chai, J., Li, W., Han, A., Chen, L., Xue, D., and Shi, Y. (2004). Structural, biochemical, and functional analyses of CED-9 recognition by the proapoptotic proteins EGL-1 and CED-4. Mol. Cell 15, 999–1006. Abstract Article
Yang, X., Chang, H.Y., and Baltimore, D. (1998). Essential role of CED-4 oligomerization in CED-3 activation and apoptosis. Science 281, 1355–1357. Abstract Article
Yuan, J., and Horvitz, H.R. (1992). The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development 116, 309–320. Abstract
Yuan, J., Shaham, S., Ledoux, S., Ellis, H.M., and Horvitz, H.R. (1993). The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75, 641–652. Abstract Article
Yuan, J.Y., and Horvitz, H.R. (1990). The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev. Biol. 138, 33–41. Abstract Article
Zarkower, D., and Hodgkin, J. (1992). Molecular analysis of the C. elegans sex-determining gene tra-1: a gene encoding two zinc finger proteins. Cell 70, 237–249. Abstract Article
Zhou, Z., Caron, E., Hartwieg, E., Hall, A., and Horvitz, H.R. (2001). The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev. Cell 1, 477–489. Abstract Article
Zhou, Z., Hartwieg, E., and Horvitz, H.R. (2001). CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104, 43–56. Article
*Edited by Geraldine Seydoux and James R. Priess. Last revised April 11, 2005. Published October 06, 2005. This chapter should be cited as: Conradt, B. and Xue D. Programmed cell death (October 06, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.32.1, http://www.wormbook.org.
Copyright: © 2005 Barbara Conradt and Ding Xue. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: barbara.conradt@dartmouth.edu or ding.xue@colorado.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.