 Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The morphogenesis of the C. elegans embryo is largely controlled by the development of the epidermis, also known as the hypodermis, a single epithelial layer that surrounds the animal. Morphogenesis of the epidermis involves cell-cell interactions with internal tissues, such as the developing nervous system and musculature. Genetic analysis of mutants with aberrant epidermal morphology has defined multiple steps in epidermal morphogenesis. In the wild type, epidermal cells are generated on the dorsal side of the embryo among the progeny of four early embryonic blastomeres. Specification of epidermal fate is regulated by a hierarchy of transcription factors. After specification, dorsal epidermal cells rearrange, a process known as dorsal intercalation. Most epidermal cells fuse to generate multinucleate syncytia. The dorsally located epidermal sheet undergoes epiboly to enclose the rest of the embryo in a process known as ventral enclosure; this movement requires both an intact epidermal layer and substrate neuroblasts. At least three distinct types of cellular behavior underlie the enclosure of different regions of the epidermis. Following enclosure, the epidermis elongates, a process driven by coordinated cell shape changes. Epidermal actin microfilaments, microtubules, and intermediate filaments all play roles in elongation, as do body wall muscles. The final shape of the epidermis is maintained by the collagenous exoskeleton, secreted by the apical surface of the epidermis.
Copyright: © 2005 Andrew D. Chisholm and Jeff Hardin. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Morphogenesis is the development of form, of tissues, of organs, and of organisms. This chapter describes the major morphogenetic processes of the mid-to-late C. elegans embryo, focusing on the epidermis and its interactions with underlying neurons and muscle. The morphogenetic movements of early gastrulation are described in Gastrulation in C. elegans . Morphogenesis of post-embryonic organs such as the gonad, vulva, and male tail, is described in the Sex determination section of WormBook.
Our description of embryonic morphogenesis centers on the epidermis, as this tissue plays the major role in determining the shape of the animal. (“Epidermis” refers to the outer cellular layer of the organism. In older literature and in WormAtlas the epidermis is named hypodermis, although the terms are synonymous in C. elegans. To avoid needless worm-specific terminology we will use “epidermis”). Morphogenesis of the epidermis involves both autonomously generated changes in epidermal cell shape and position, and interactions with internal tissues, including the developing nervous system and body wall muscles. Molecules required for epidermal morphogenesis thus include components of the epidermal cytoskeleton, epidermal cellular junctions, cell signaling pathways operating between the epidermal cells and underlying tissues, and of the extracellular matrix. More detailed accounts of these molecules can be found in the Cell biology section of WormBook.
At the time of hatching, the C. elegans epidermis is almost entirely composed of syncytia formed by fusions of epidermal cells. The process and mechanism of cell fusion is described by Podbilewicz (see Cell fusions). Mutants defective in cell fusion, such as eff-1, display aberrant elongation of the epidermis (Mohler et al., 2002), indicating that cell fusion is required for normal epidermal morphogenesis; the mechanistic basis for this requirement has not been fully elucidated.
In this review we follow the development of the epidermis during embryogenesis, focusing on processes and tissue interactions required for its morphogenesis. Our discussion of genes or molecules involved in these developmental processes is selective, and focuses on pathways whose role in the morphogenetic process has been analyzed in most detail. We begin with the specification of the major epidermal fates, then describe the morphogenetic processes of epidermal enclosure, dorsal intercalation, and elongation. Both enclosure and elongation exemplify organogenesis processes in that they involve interaction with underlying tissues: ventral neuroblasts are required for epidermal enclosure, whereas body muscles are required for epidermal elongation. We conclude with the role of the extracellular matrix, including the cuticle, in embryonic epidermal development.
The anatomy of the mature epidermis is described by Hall (see the Anatomy section of WormBook). In this chapter we focus on the morphogenesis of the major epidermal cells, as defined by Gendreau et al. (Gendreau et al., 1994) and modified by Page et al. (Page et al., 1997). The 71 major epidermal cells form most of the epidermis; an additional 11 minor epidermal cells form epidermal syncytia at the extreme head and tail.
The major epidermal cells are large cells that are generated after the 9th round of embryonic cell divisions, 220-240 minutes after first cleavage; at this stage the embryo is composed of ~365 cells. The major epidermal cells are derived from four of the 12-cell stage blastomeres: ABarp, ABpla, ABpra, and C. These blastomeres also give rise to other cell types: the AB blastomeres generate neuronal cells and C gives rise to body muscle. In general, epidermal cells arise piecemeal within these lineages; only at the 7th round of cleavages do exclusively epidermal precursors arise. The major epidermal precursors are located on the dorsal surface of the early embryo, and thus the major epidermal cells arise as a group on this surface. The minor epidermal cells are smaller and born later, after the 10th round of division. Once the terminal divisions that produce the epidermis occur, three major groups of cells are evident within the epidermis: (1) dorsal epidermal cells arise as two rows of ten cells each along the anterior-posterior axis, straddling the dorsal midline; (2) two more lateral rows of “seam” epidermal cells flanking the dorsal cells; and (3) ventral epidermal cells lying at the extreme lateral edges of the epidermis, ultimately migrating to meet at the ventral midline.
Soon after their birth, the major epidermal cells express and begin to localize epithelial markers, such as the AJM-1 protein. Formation of an epidermal sheet thus begins within a few minutes of the birth of the major epidermal cells (McMahon et al., 2001). The initial specification of epidermis requires the GATA family Zinc finger transcription factor ELT-1. elt-1 null mutants completely fail to undergo morphogenesis and contain excess neurons and muscles instead of epidermis. ELT-1 is expressed from about the 28-cell stage in cells that will give rise to, among other things, the major epidermal cells (Page et al., 1997). It is likely that ELT-1 directly activates the transcription of at least two other transcription factors, the zinc finger protein LIN-26 and the GATA family protein ELT-3.
LIN-26 expression begins at the 100-cell stage in precursors of major epidermal cells; LIN-26 expression is continuous in these lineages. LIN-26 acts to maintain epithelial aspects of the epidermal fate; in lin-26 null mutants epidermal fates are specified but abnormal; for example, epidermal cells fail to develop a normal apical-basal polarity. lin-26 mutants display variable failures in morphogenesis, including defects in epidermal enclosure and elongation. Epidermal cells in lin-26 mutants eventually become necrotic (Labouesse et al., 1994).
ELT-3 is activated in all post-mitotic epidermis, except the seam cells, until the 2.5-fold stage; ELT-3 expression then declines until it is reactivated in a second phase in the valve cells at either end of the gut (Gilleard et al., 1999). An elt-3 null mutation does not cause detectable effects on embryogenesis, suggesting that ELT-3 functions redundantly with other epidermal regulators (Gilleard and McGhee, 2001).
LIN-26 and ELT-3, and perhaps other proteins, likely regulate genes required for the expression of epidermal fates. Such genes must include those required generally in all epithelial cells, and those required specifically in the epidermis, such as cuticle components. Ectopic expression of LIN-26 induces expression of the general epithelial markers AJM-1, DLG-1, and CHE-14, but not of genes specific to epidermal cells (Quintin et al., 2001). However, AJM-1 and CHE-14 expression is normal in lin-26 null mutant epidermal cells. Thus, LIN-26 is sufficient, but not necessary, for expression of these epithelial markers, suggesting that multiple factors act combinatorially to completely specify the epidermal fate.
Ventral neuroblasts form the substrate for later enclosure movements of the epidermis, and so their development is considered here. Ventral neuroblasts themselves undergo morphogenetic movements together known as ventral cleft closure (Movie 1). During gastrulation precursors of the gut, pharynx, and body muscles ingress on the ventral surface of the developing embryo (see Gastrulation in C. elegans ). These ingression movements create a depression on the ventral surface, known as the ventral cleft (Figure 1 and Figure 2; Sulston et al., 1983). The ventral cleft is surrounded by neuroblasts derived from ABplp and ABprp in the posterior and ABalp and ABarp in the anterior. These ventral neuroblasts move en masse from more lateral positions towards the ventral midline between 230 and 290 minutes, causing the ventral cleft to disappear by about 290 minutes, roughly an hour before epidermal enclosure begins. Ventral neuroblast movements begin at the posterior cleft and progress towards the anterior. The rate and timing of individual neuroblast movements have not been examined in detail, nor is it yet known if specific cells or cell groups play different roles in the movement. The ventral neuroblasts undergo their last major round of divisions between 250 and 310 minutes.

Figure 1. Developmental timing of morphogenetic events. Developmental landmarks are shown with representative Nomarski Differential Interference Contrast (DIC) images of embryos at corresponding stages, based on Figure 4 of Sulston et al. 1983 (Sulston et al., 1983). Developmental times are relative to the division of the zygote (P0), in minutes at 20°C; rates at 25° are 1.2 times faster than at 20°C; rates at 15°C are 1.5 times slower than at 20°C. Fertilization occurs ~45 minutes before P0 divides. The stage termed ‘lima bean’ has not been precisely defined but is roughly equivalent to the period of epidermal enclosure when the ventral side of the embryo first becomes visibly concave. Note that unequal growth along the anteroposterior axis forces the embryo to turn within the eggshell between 350 and 400 minutes so that either the left or right side is uppermost, when mounted for observation; images before turning are either ventral or dorsal views, whereas afterwards they are left or right lateral views. All laterally viewed embryos in this and subsequent figures are oriented with anterior to the left, posterior to the right.
Movie 1. Ventral cleft closure in a wild-type embryo visualized using Nomarski microscopy. Anterior to the left, ventral view. Ventrolateral neuroblasts move towards the ventral midline and close the cleft from posterior to anterior. Frames were acquired at 60 sec intervals. Original movie footage courtesy of H.N. Cinar and D. Patel (unpublished).
Defects in the movement of ventral neuroblasts are seen in animals defective in any of several cell signaling pathways, including Eph receptor/ephrin, PTP-3/LAR RPTP (Leukocyte Common Antigen Related Receptor Protein Tyrosine Phosphatase), and semaphorin-2A/MAB-20 (Chin-Sang et al., 1999; Chin-Sang et al., 2002; George et al., 1998; Harrington et al., 2002; Roy et al., 2000; Wang et al., 1999). Such defects result in an enlarged or persistent ventral cleft; if the ventral cleft is not closed by the time of epidermal enclosure, enclosure movements can be blocked. The cellular defects of the neuroblasts in these mutants have not yet been elucidated. Loss of function in the SCAR/WAVE gene wve-1 causes drastic defects in ventral neuroblast organization (Withee et al., 2004), suggesting that actin-based motility may be important for ventral neuroblast movement. However, abnormalities in actin distribution in ventral neuroblasts have not yet been observed in such mutants, possibly reflecting the limited resolution of available reagents. Ventral cleft closure is also defective in some neuronal cell fate mutants such as unc-62 and pal-1 (Van Auken et al., 2002). Mutations in the pha-4 gene, which block pharynx specification, cause failure of ingression of pharyngeal precursors and later defects in epidermal enclosure and elongation, presumably reflecting a defective substrate (Mango et al., 1994).
Interestingly, null mutations in several genes cause slight delays in neuroblast migration, detectable by 4-D timelapse microscopy, that result in low penetrance epidermal enclosure defects. Such genes include the atypical ephrin efn-4, the Wiskott-Aldrich Syndrome Protein ortholog wsp-1 (Withee et al., 2004), and the C. elegans Kallmann syndrome ortholog kal-1 (Rugarli et al., 2002; M.L. Hudson and A.D.C., unpublished results). These subliminal defects in neuroblast migration nevertheless appear to sensitize the embryo to loss of function in other pathways acting in neuroblast movement, as illustrated by the extreme synergistic effects of loss of function mutations in efn-4 and vab-1 (Chin-Sang et al., 2002).
The nature of the interaction between migrating epidermal cells and the neuronal substrate remains poorly understood. Epidermal enclosure may require a uniform substratum of cells; if this substrate is disrupted by a persistent ventral cleft, epidermal cells may be physically unable to enclose the embryo. Although it is possible that specific substrate cells may play instructive roles in guiding the movement of the overlying epidermis, laser killing experiments did not reveal individual cells that could play such roles (Williams-Masson et al., 1997).
The preceding sections described the processes that lead to a dorsal sheet of epidermal cells and a ventral surface of substrate neuroblasts. Before epidermal enclosure begins, two rows of dorsal epidermal cells engage in a striking rearrangement, known as dorsal intercalation (Movie 2 and Movie 3), to form a single row across the dorsal midline; intercalation begins approximately 10 minutes after the terminal divisions of epidermal precursors. During the first stages of intercalation, dorsal cells become wedge shaped, with their pointed tips oriented toward the dorsal midline. Dorsal cells then interdigitate between their contralateral neighbors, eventually elongating until their extending edges make contacts with seam cells on the opposite side of the embryo (Sulston et al., 1983; Williams-Masson et al., 1998; Figure 3). The nuclei of intercalating cells also migrate contralaterally (Sulston et al., 1983; Williams-Masson et al., 1998); however, nuclear migration is not required for successful intercalation (Starr et al., 2001).
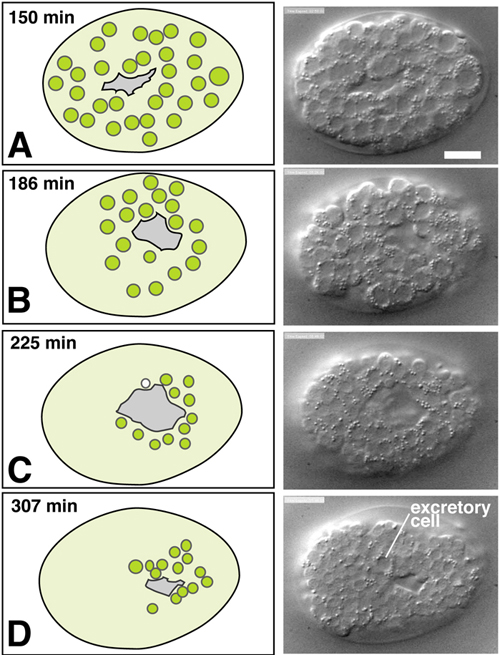
Figure 2. Ventral cleft closure. Nomarski DIC images (courtesy H.N. Cinar and D. Patel) and schematics of ventral neuroblast positions during closure of the ventral cleft in the wild type; see also Movie 1. The left column includes schematics of ventral views of embryos with the migrating neuroblasts shown in green. The right column includes DIC images or embryos at the corresponding stage. Although the movements of individual cells during ventral cleft closure have not been fully described, the presence of a ventral cleft is well defined and its duration can be quantitated (M. L. Hudson and A.D.C., unpublished results). Scale, 10 μm.
Movie 2. Dorsal intercalation in a wild-type embryo visualized using Nomarski microscopy (Williams-Masson et al., 1998). Cells become wedge-shaped as they begin intercalation, and eventually adopt a ladder-like arrangement straddling the dorsal midline. Anterior to the left, dorsal view. Frames were acquired at 30 sec intervals. Original movie footage courtesy of E. Williams-Masson.
Movie 3. Dorsal intercalation in a wild-type embryo visualized using a dlg-1::gfp translational fusion (Koppen et al., 2001). Frames were acquired at 5 min. intervals. Original multiphoton movie footage courtesy of M. Köppen.
The mechanisms by which dorsal cells rearrange is not well understood. Pharmacological studies indicate that both actin microfilaments and microtubules are required (Williams-Masson et al., 1998), but the precise role each cytoskeletal system plays is unclear. Based on serial reconstructions of TEM thin sections (Williams-Masson et al., 1998) and on dynamic imaging of a cytoplasmic GFP reporter (Heid et al., 2001), dorsal cells extend basolateral membrane protrusions in the direction of rearrangement. After an initial period of protrusive activity at both their lateral and medial borders, dorsal cells extend highly polarized protrusions that extend far beyond the position of the overlying apical junctions (Figure 5a-c; Movie 4). Such activity is similar to the "monopolar" activity of some rearranging cells in the Xenopus neural plate (Elul and Keller, 2000) and the ascidian notochord (Munro and Odell, 2002), and presumably allows dorsal epidermal cells to change position. What molecules are required for the extension of these basolateral protrusions, and what, if any, force they exert is unknown. Regardless of the mechanism, several lines of evidence suggest that an individual dorsal cell may be able to effect the cell-shape changes required for intercalation in a cell-autonomous fashion. Removal of the posterior portion of the dorsal epidermal array by ablation of the C blastomere, neighboring seam cells, or underlying muscle does not disrupt intercalation of the remaining cells (Heid et al., 2001; Williams-Masson et al., 1998).
Little is known about the molecular control of dorsal intercalation. The C2H2 Zinc finger transcription factor DIE-1 is required for completion of intercalation, but not for the initial polarization of intercalating cells towards the dorsal midline (Heid et al., 2001). Mosaic loss of DIE-1 in dorsal cells results in cell autonomous failure to complete intercalation, suggesting that DIE-1 is required in all intercalating cells (Heid et al., 2001). Transcriptional targets of DIE-1 have not yet been identified. Other genes are likely required for the correct patterning of the dorsal epidermis. These include the functionally redundant T box proteins, TBX-8 and TBX-9. Loss of function in either tbx-8 or tbx-9 results in occasional defects in the positioning of some dorsal cells, whereas tbx-8 tbx-9 double mutants or double RNAi embryos display severe disruption of the positioning and/or morphogenesis of the dorsal epidermis (Andachi, 2004; Pocock et al., 2004). Whereas die-1 mutant dorsal cells appear correctly patterned and in the correct positions but fail to complete intercalation, dorsal cells in tbx-8 tbx-9 double mutants can complete intercalation even when they initiate it from an incorrect position (Pocock et al., 2004), suggesting that DIE-1 and TBX-8/TBX-9 regulate different phases of the process.
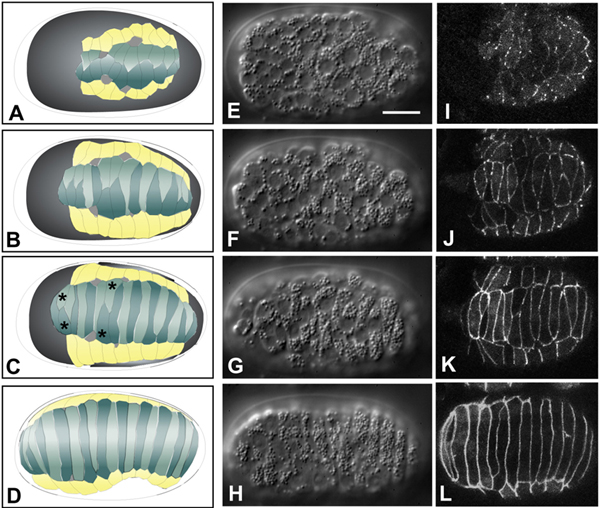
Figure 3. Dorsal intercalation. Schematics (left column, A-D), Nomarski DIC images (middle column, E-H; dorsal views; see also Movie 2) and DLG-1::GFP images highlighting epidermal cell boundaries (right column, I-L; dorsal view; see also Movie 3) of dorsal intercalation. Different embryos, at corresponding developmental stages, are shown in middle and right columns. Scale bar = 5 μm. For details on DLG-1, which localizes to apical junctions in epithelial cells in C. elegans, (see Köppen et al. (2001) and Epithelial junctions and attachments. Dorsal cells are shown in teal in the left-hand column, seam epidermal cells are shown in yellow, and deirids are shown in white. Dorsal cells born on the right side of the embryo are depicted in a lighter color than those born on the left. For clarity, ventral epidermal cells and non-epidermal tissues are not shown. The last two pairs of dorsal epidermal cells to intercalate have been termed "pointer cells" (Williams-Masson et al., 1998), and serve as fiduciary markers for the progress of intercalation in wild-type embryos (C, asterisks). Nomarski images in this figure and Figure 4 courtesy of E. Williams-Masson; DLG-1::GFP images courtesy of M. Köppen.
Movie 4. Visualizing protrusive activity in wild-type embryos during dorsal intercalation using an lbp-1::gfp transcriptional reporter. Frames were acquired at 5 min. intervals. To suppress cell fusion, the strain was made homozygous for the eff-1(oj55) allele (Heid et al., 2001). Note the basolateral protrusions extended by the dorsal epidermal cells as they intercalate. The bright ovals are the nuclei of dorsal epidermal cells, which undergo a contralateral migration as intercalation proceeds. Anterior is to the left. Original spinning disc confocal movie footage courtesy of R. King.
Movie 5. Ventral enclosure in a wild-type embryo visualized using Nomarski microscopy (Williams-Masson et al., 1997). Anterior to the left, ventral view. Frames were acquired at 2 min. intervals. Original movie footage courtesy of M. Köppen.
Movie 6. Ventral enclosure in a wild-type embryo visualized using a dlg-1::gfp translational fusion (Koppen et al., 2001). Frames were acquired at 5 min. intervals. Original multiphoton movie footage courtesy of M. Köppen.
Ventral enclosure (Movie 5, Movie 6 and Movie 7; Figure 4), the process by which ventral epidermal cells migrate toward the ventral midline to encase underlying cells in an epithelial monolayer, ensues shortly after dorsal intercalation begins (Figure 1). Based on 4-D Nomarski and multiphoton imaging, ventral enclosure occurs in two phases. First, two pairs of anterior, or ‘leading’ cells extend large protrusions as they migrate towards the ventral midline (Figure 5d-f; Movie 6). The anterior pair of leading cells are the two ventral cells that will later fuse to the hyp6 syncytium; the posterior pair will eventually become part of hyp7. The migratory activity of these cells is essential for enclosure, since ventral enclosure can be completely blocked by the laser inactivation of these two pairs of cells (Williams-Masson et al., 1997). In these experiments the leading cells were irradiated with laser power sufficient to prevent their movement, but not sufficient to kill them. In the second step, posterior ventral cells become wedge shaped and elongate toward the ventral midline, lining a ‘pocket’ at the ventral midline (Figure 4). Laser inactivation of a significant fraction of the ventral pocket cells also results in failure of enclosure; the elastic recoil of the leading edge in such experiments further suggests that the pocket is under mechanical tension. Ultimately, ventral epidermal cells make bilaterally symmetric contacts, thereby establishing new junctional connections at the ventral midline. The extreme anterior epidermis then completes the enclosure of the embryo by a sheet of epidermis (Figure 4).

Figure 4. Epidermal enclosure. Schematics (left column, A-D), Nomarski images (middle column, E-H, ventral view; see also Movie 5) and DLG-1::GFP images (right column, I-L, ventral view; see also Movie 6) of ventral enclosure. Different embryos, at corresponding developmental stages, are shown in middle and right columns. Scale bar = 5 μm. Ventral cells are shown in pink in the left-hand column; seam epidermal cells in yellow, and dorsal cells, which wrap around the tail, in teal. Neuroblasts and other internal cells are depicted in gray. The first two pairs of ventral cells to reach the ventral midline are termed "leading cells" (Williams-Masson et al., 1997), which extend long protrusions towards the midline (B). After the leading cells make contact at the ventral midline, cells posterior to these cells, termed "pocket cells" (Williams-Masson et al., 1997), reach the midline (C). Finally, the anterior epidermal cells enclose the head (D).
Movie 7. Ventral enclosure in a wild-type embryo visualized using a dlg-1p::gfp transcriptional reporter (Firestein and Rongo, 2001). Frames were acquired at 2 min. intervals. Original spinning disc confocal movie footage courtesy of M. Sheffield.
Pharmacological studies indicate that both the actin and microtubule cytoskeletal systems are required for enclosure (Williams-Masson et al., 1997). The requirement for the latter is consistent with enclosure defects observed in evl-20/ARL2 mutants, which also show defects in microtubule organization (Antoshechkin and Han, 2002).

Figure 5. Protrusive activity during morphogenesis. Both dorsal (top row, A-C) and ventral (bottom row, D-F) epidermal cells display protrusive activity as they migrate. (A) A schematic of cell wedging during dorsal intercalation, corresponding to Figure 3B. (B) An embryo expressing an lbp-1::gfp transcription reporter to mark epidermal cells (Heid et al., 2001), imaged using spinning disk confocal microscopy (courtesy of R. King; see also Movie 4). Mosaicism has resulted in expression only in the righthand cohort of posterior dorsal epidermal cells. Rearranging cells extend long protrusions in the direction of migration (arrow). (C) A schematic showing the relationship between such basolateral protrusions and rearrangement of dorsal cells (here and in F, anterior at the top). Protrusions lie basal to the apical junctional domain (pink). In addition, although it is not understood, wedging must presumably remodel junctional proteins as they rearrange (red; see Hardin and Walston (2004) for detailed discussion). (D) A schematic of leading cell migration during ventral enclosure, corresponding to Figure 4B. (E) An embryo expressing a dlg-1::gfp transcriptional reporter (Firestein and Rongo, 2001a) visualized using spinning disk confocal microscopy (courtesy of M. Sheffield; see also Movie 7). Migrating leading cells extend long protrusions in the direction of migration (arrows); in time-lapse sequences, pocket cells can also be observed with shorter protrusions. Scale bar = 5 μm. (F) A schematic showing the relationship between leading cell protrusions and the underlying cellular substratum of neuroblasts (anterior at the top). Protrusions make contact via cadherin/catenin complex proteins on their surfaces (green). F-actin is abundant within the cytoplasm of extending protrusions and may connect to cadherin complex proteins (“filopodial priming”; Raich et al., 1999a).
Several permissive requirements for leading cell migration and adhesion have been identified. First, Rac-dependent modulation of the actin cytoskeleton, presumably operating through the Arp2/3 complex, is required for the onset of migration. Progeny of ced-10/Rac mutant mothers (Soto et al., 2002; Lundquist et al., 2001), as well as embryos depleted of components of the Arp2/3 complex (Sawa et al., 2003; Severson et al., 2002), and the Rac interacting proteins GEX-2/Sra-1 and GEX-3/HEM2/NAP1/KETTE (Gex = gut on the exterior) display a highly disorganized epidermis that is largely unable to undergo ventral migration (Soto et al., 2002). CED-10 (Chen et al., 1996), GEX-2, and GEX-3 (Soto et al., 2002) all localize to the periphery of epidermal cells near apical junctions, suggesting that they are important for maintaining epidermal cell shape and protrusive activity. The Scar/Wave protein GEX-1/WVE-1 likely acts as a CED-10 effector in this pathway to activate the Arp2/3 complex, based on similarity of phenotype (M. Soto, personal communication; M. Sheffield and J. Hardin, unpublished). This same pathway may also be required for successful dorsal intercalation, although further analysis will be required to confirm this (M. Sheffield and J. Hardin, unpublished).
Once leading cell migration is underway, regulation of leading cell protrusive activity is important for continued migration. The WASP family protein WSP-1 and the Ena/VASP family protein, UNC-34, are likely to be important during this process: tissue-specific knockdown of wsp-1 function in epidermal cells (Sawa et al., 2003) or simultaneous depletion of UNC-34 and WSP-1 (Withee et al., 2004) results in enclosure defects. Since both WASP and Ena/VASP family proteins regulate activation of the Arp2/3 complex and filament dynamics at the plus ends of actin filaments, respectively, these likely exert their effects on enclosure by regulating the polymerization of actin at the free edges of the enclosing epidermis. Cell signaling, in part mediated via inositol trisphosphate (IP3), also appears to regulate leading cell migration. Offspring of hermaphrodites carrying a weak mutation in the single C. elegans IP3 receptor gene, itr-1, display reduced protrusive activity at the leading edge and a variety of enclosure defects (Thomas-Virnig et al., 2004).
Another requirement for successful leading cell function is the cadherin/catenin junctional complex. Leading cells in embryos lacking both maternal and zygotic contributions of any of the components of the cadherin/catenin complex (HMR-1A/cadherin, HMP-2/β-catenin, or HMP-1/α-catenin; see The cadherin superfamily and Epithelial junctions and attachments for further information) can extend protrusions, but these protrusions fail to make stable adhesive contacts at the ventral midline (Movie 8). Analysis of HMP-1::GFP distribution in living embryos indicates that leading cell protrusions accumulate cytoplasmic HMP-1, which is rapidly recruited to nascent junctions upon their contact at the midline, a process that has been termed "filopodial priming" (Raich et al., 1999).
Movie 8. Loss of cadherin function specifically disrupts leading cell adhesion. In this example, a hmr-1(RNAi) embryo expressing ajm-1::gfp ruptures during ventral enclosure (Raich et al., 1999). As ventral epidermal cells approach the ventral midline, the pocket cells migrate in advance of the leading cells. Epidermal cells that fail to form junctions at the ventral midline retract towards the dorsal surface of the embryo. Anterior to the left, ventral view. Frames were acquired at 5 min. intervals.
Whereas the migratory behavior of leading cells clearly leads to their ventral migration, how the ventral pocket closes is less certain. Actin is concentrated at the ventral tips of pocket cells, which suggests that the ventral pocket may close via a multicellular purse-string mechanism (Williams-Masson et al., 1997). However, multiphoton imaging has shown that pocket cells also extend short protrusions towards the ventral midline as they migrate (Thomas-Virnig et al., 2004). Both filopodial protrusions and a robust actin cable have been observed during Drosophila dorsal closure; the former are presumed to aid bilateral cell-cell matching and the latter contributes forces for closure (Jacinto et al., 2002). Similar mechanisms may operate during ventral pocket closure in C. elegans. That regulation of protrusive contacts between pocket cells is important during pocket closure is suggested by defects in mutants for mab-20/Ce-sema-2a, which encodes a secreted Semaphorin. mab-20 mutants display ectopic contacts between adjacent pocket cells during enclosure (Roy et al., 2000). Semaphorins typically mediate repulsive guidance cues in other contexts (Pasterkamp and Kolodkin, 2003); MAB-20 may limit adhesion between adjacent pocket cells as they migrate by preventing ectopic protrusive activity.
The enclosure of the head epidermis (defined as the syncytia hyp1-5) has not been extensively characterized. Correct specification of head epidermal cells is likely necessary for head enclosure, as loss of function in the homeobox genes ceh-13/labial or ceh-43/Distal-less results in ruptures of head epidermis (Aspock and Burglin, 2001; Brunschwig et al., 1999). Based on SEM, multiphoton and spinning disk confocal imaging, the extreme anterior epidermal cells do not produce long, anterior-directed protrusions, although some epidermal cells that contribute to the hyp3 and hyp4 syncytia display long protrusions as they migrate (J. Hardin, unpublished observations). As with ventral epidermal cells in the trunk, semaphorin-mediated signaling may regulate contacts between adjacent ventral cells. In this case, the transmembrane semaphorin, SMP-1/Ce-sema-1a, may be redundant with MAB-20. smp-1 mutants display a "displaced mouth" phenotype, indicative of defects in the morphogenesis of hyp3; these defects are exacerbated in mab-20; smp-1 double mutants (Ginzburg et al., 2002).
Immediately after the epidermis is enclosed, elongation converts the bean-shaped embryo into the elongated shape of the worm (Movie 9). In our discussion of elongation we focus on the epidermis, as this tissue is thought to provide the driving force for the elongation movement. Based on analysis of pharyngeal specification mutants (Mango et al., 1994) and laser killing experiments (Junkersdorf and Scheierenberg, 1992), the pharynx and intestine may not be required for the embryo to elongate. However, at least one internal tissue (body muscle) is essential for full elongation of the epidermis.
Movie 9. Elongation in a wild-type embryo imaged using Nomarski microscopy. Frames were collected at 60 sec intervals. Anterior to the left, lateral view. Original movie footage courtesy of E. Cox (Pettitt et al., 2003).
Epidermal elongation begins ~350 minutes after first cleavage and is largely complete by 600 minutes (Figure 1). During elongation, the embryo reduces its circumference by a factor of three and increases in length by a factor of four. Elongation after the 1.5-fold stage (430 minutes) proceeds at ~50 μm/hour until ~600 minutes (McKeown et al., 1998). The rate of elongation is highly reproducible and late C. elegans embryos are developmentally staged by their extent of elongation (Figure 1).
Elongation of the embryo reflects the elongation of epidermal cells along the anterior-posterior axis (Priess and Hirsh, 1986). The apical surfaces of epidermal cells shorten around the animal’s circumference while elongating in the orthogonal direction, along the anterior-posterior axis. Laser killing experiments have demonstrated that the integrity of the epidermal sheet is essential for elongation (Priess and Hirsh, 1986). An essential role for underlying muscle cells in later elongation has been shown by genetic analysis (see below).
The onset of elongation involves several processes. First, both actin microfilaments and microtubules within dorsal and ventral epidermal cells become highly organized in a striking circumferential pattern, in association with the apical plasma membrane (Costa et al., 1997; Costa et al., 1998; Priess and Hirsh, 1986; Williams-Masson et al., 1997). Both filament systems are required for elongation: treatment with the microfilament inhibitor cytochalasin D prevents elongation entirely, and treatment with the microtubule inhibitor nocodazole results in disorganized elongation. Remarkably, the effects of cytochalasin are reversible (Priess and Hirsh, 1986). Although the mechanisms by which circumferential actin filament bundles (CFBs) form in such an ordered fashion is unknown, their assembly is transient; actin filaments do not align until near the end of ventral enclosure, and they do not persist after elongation is complete (Costa et al., 1997; Williams-Masson et al., 1997).
Genetic analysis has demonstrated that actomyosin-based contraction of epidermal cells is crucial for elongation. Loss of function of the C. elegans homologue of Rho-binding kinase (LET-502) results in reduced elongation; conversely, loss of function of the myosin phosphatase, MEL-11, results in "hypercontraction", as local regions of excessive constriction occur. Null mutants for let-502 and mel-11 suppress one another, indicating that LET-502 and MEL-11 act antagonistically during elongation. The relatively normal elongation in let-502 mel-11 double mutants indicates that this pathway is redundant with additional pathways in regulating elongation; components of such parallel pathways include the phosphatase FEM-2, better known for its function in sex determination (see the Sex determination section of WormBook; Piekny et al., 2000; Wissmann et al., 1999; Wissmann et al., 1997).
Based on the expression patterns of these regulatory proteins (see below), it is thought that the actomyosin-based contractile machinery is predominantly active in the lateral seam cells. Seam cells may transmit contractile forces to dorsal and ventral cells via CFBs and cadherin-catenin junctions; the dorsal and ventral cells may not actively constrict, but may passively respond to seam-generated forces. However, components of dorsal and ventral epidermal cell cytoskeleton become essential for epidermal elongation after the two fold stage; although this requirement is not fully understood, it suggests that dorsal and ventral cells may actively reorganize their cytoskeleton in later elongation.
Both LET-502 and MEL-11 are expressed at high levels in the cytoplasm of seam cells; when elongation starts, MEL-11 becomes confined to cell-cell junctions where it can no longer inhibit the contractile apparatus (Piekny et al., 2003). Moreover, LET-502 is coextensive with MLC-4, a myosin regulatory light chain that is expressed at high levels in seam cells (Shelton et al., 1999). mlc-4 is required zygotically for elongation (Shelton et al., 1999), making MLC-4 a likely target of LET-502. Together, these data suggest that by acting on MLC-4, LET-502 promotes actomyosin-mediated contraction, whereas MEL-11 mediates relaxation: in seam cells, contractile forces predominate and the cells elongate. Two non-muscle myosin heavy chains, NMY-1 and NMY-2 have partially redundant roles in elongation and are presumably both regulated by MLC-4 (Piekny et al., 2003).
The linkage of CFBs to adherens junctions is critical for converting the forces of contraction into coordinated elongation across the entire epidermis. Mutations in genes encoding core proteins of the cadherin-catenin complex (hmp-1, hmp-2, and hmr-1) or associated proteins result in elongation failure or abnormalities due to detachment of CFBs. Whereas absence of maternally and zygotically contributed cadherin-catenin complex proteins result in enclosure failure (often the Hammerhead (Hmr) phenotype; see above), zygotic hmp-1 and hmp-2 mutants display a milder Humpback (Hmp) phenotype, in which embryos enclose but form abnormal bulges when they attempt to elongate (Costa et al., 1998). In Hmp mutants CFBs visibly detach from junctions. Based on its genetic interactions with a weak loss of function allele of hmp-1(fe4), JAC-1/p120ctn positively modulates cadherin-catenin function in C. elegans but is not itself essential (Pettitt et al., 2003). The claudin-like putative four-pass transmembrane protein VAB-9 also localizes to the adherens junctional domain and requires HMR 1 for its localization (Simske et al., 2003). vab-9 null mutants have elongation and body-shape abnormalities and perturbed CFB organization (Simske et al., 2003; Figure 6).
The epidermal spectrin cytoskeleton is essential for epidermal elongation. Loss of function in the sole C. elegans α-spectrin, SPC-1, causes embryos to arrest in mid-elongation (Norman and Moerman, 2002). C. elegans expresses two β-spectrins, the conventional βG-spectrin UNC-70 (Hammarlund et al., 2000; Moorthy et al., 2000) and the βH-spectrin SMA-1. Null mutants in sma-1 (for Small) display slower elongation (McKeown et al., 1998) and variable defects in the organization of actin in seam cells (Praitis et al., 2005). UNC-70 does not appear to be required individually for elongation but may account for the residual elongation of sma-1 null mutants (Moorthy et al., 2000). SMA-1 is associated with the apical membrane in dorsal and lateral epidermis (Praitis et al., 2005), and the organization of CFBs is mildly abnormal in both sma-1 and spc-1 mutants (McKeown et al., 1998; Norman and Moerman, 2002). This suggests that a SMA-1/SPC-1 complex could stabilize actin filaments at the apical surface of epidermal cells, particularly in seam cells as they contract.
Elongation beyond the two-fold stage appears to require additional molecular mechanisms in addition to the components described above (Ding et al., 2004). The first evidence that later stages of elongation may differ mechanistically from the earlier stages came from analysis of mutants affecting muscle function. Mutations causing complete absence of muscle function result in failure to elongate beyond the two-fold stage (the Pat, paralyzed arrest at two-fold, phenotype; Williams and Waterston, 1994). In such mutants elongation appears to proceed at a normal rate until the two-fold stage when it ceases.
The mechanism by which muscle function promotes epidermal cell shape change is not known. It is known that muscles induce and localize hemidesmosome-containing trans-epidermal attachment structures (‘fibrous organelles’) within the epidermal cytoskeleton (Francis and Waterston, 1991; Hresko et al., 1994); for a detailed description of the molecules of attachment structures, see Epithelial junctions and attachments. Attachment structures can be regarded as analogous to tendons, transmitting the force of muscle contraction to the cuticular exoskeleton. Components of these attachment structures, such as Myotactin/LET-805 (Hresko et al., 1999), Plectin/VAB-10A (Bosher et al., 2003), Intermediate filament (IF) proteins IFB-1 and IFA-3 (Woo et al., 2004), and the novel cytoplasmic adaptor protein VAB-19 (Ding et al., 2003), are essential for epidermal elongation past the two-fold stage. At present there is no evidence that IFs function in earlier elongation or morphogenesis, although this conclusion is qualified by the lack of data on the function of most C. elegans IFs.
Unlike “strong Pat” mutants, which are completely paralyzed, mutants defective in epidermal attachment structures display initially normal muscle contractions from the 1.75-fold stage, prior to detachment of muscles from the epidermis. The interdependence of muscles and epidermal attachment structures confounds efforts to determine whether the role of muscles in elongation reflects their function in formation of trans-epidermal attachments, or whether attachment structures, by allowing muscle force transduction to the epidermis, promote some additional muscle-dependent function. If the role of muscles is to induce and organize attachment structures, this begs the question of why trans-epidermal attachments (which are only found in dorsal and ventral epidermis) are required for elongation. Attachment structures likely interact with other components of the epidermal cytoskeleton, such as circumferential actin and microtubule bundles or the apical spectrin cytoskeleton. Abnormalities in the trans-epidermal attachment structures may in turn result in defects of the cytoskeletal structures required for elongation. The molecular basis for such interactions is not yet known.
The extracellular matrix basement membrane (bm) secreted by the epidermis is essential for normal elongation, although the requirements for bm components can vary. Lack of UNC-52/Perlecan causes elongation to be blocked at the two-fold stage, the ‘severe Pat’ phenotype, reflecting an essential role for perlecan in integrin-mediated assembly of the myofilament lattice in body wall muscles (Hresko et al., 1994). In contrast, lack of the α(IV) basement membrane collagens EMB-9 or LET-2 causes progressive muscle detachment and a late block in elongation at the three-fold stage (Gupta et al., 1997) (the ‘Lat’ phenotype described by Williams and Waterston, 1994). Because EMB-9 and LET-2 only localize to the regions of bm between muscle and epidermis, the requirement for bm collagens again reflects the requirement for muscle-epidermal interaction for normal epidermal elongation. Mutations affecting laminin α result in pleiotropic effects on epidermal morphogenesis that may reflect both failure to maintain epidermal apical-basal polarity and specific defects in muscle-epidermal attachment (Huang et al., 2003).
During elongation the shape of the epidermis is actively regulated by the epidermal actin cytoskeleton. At their apical surface epidermal cells secrete the extracellular embryonic sheath, whose function is not well understood. After elongation is complete, the epidermis begins to secrete the cuticle of the first stage larva. The cuticle now takes over the role of holding the epidermal cells in place. Animals lacking the cuticle collagen SQT-3 elongate normally but then fail to maintain the elongated form and retract (Priess and Hirsh, 1986). Similar late defects in epidermal morphogenesis result from loss of function in other collagens or in enzymes required for collagen processing such as the protease DPY-31 (Novelli et al., 2004). Defects in cuticle secretion also result in late-onset defects in elongation (Roberts et al., 2003).
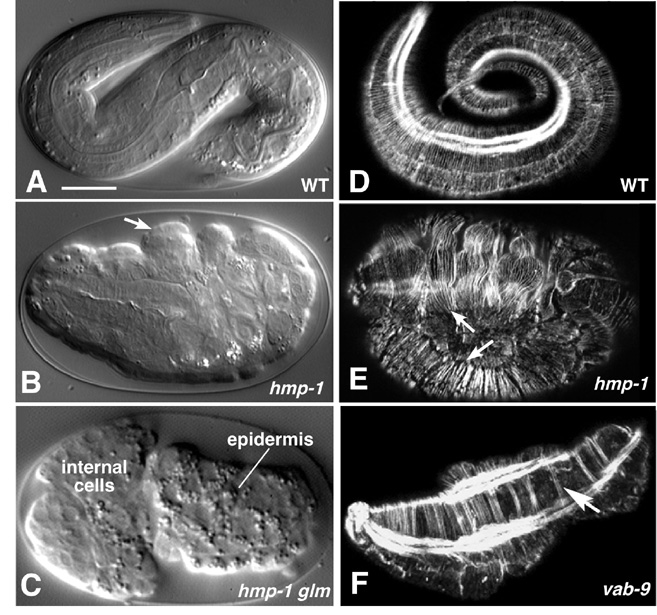
Figure 6. Requirements for cadherin-based adhesion during morphogenesis. The cadherin/catenin complex and associated proteins are required for ventral enclosure and elongation. Left column (A-C), Nomarski images; right column (D-F), phalloidin staining of F-actin imaged using confocal microscopy. (A) Wild-type embryo at approximately the three-fold stage. (B) A hmp-1(zu278) homozygote from a heterozygous mother, displaying the Hmp (Humpback) phenotype. Prominent dorsal bulges in the epidermis are visible (arrows). (C) An embryo from a hmp-1(zu278) germline mosaic mother, which therefore lacks both maternal and zygotic HMP-1, displaying the more severe Hmr (Hammerhead) phenotype. Enclosure failure in the anterior results in rupture of the embryo and ejection of internal cells through the opening in the epidermis. For a multiphoton movie of such enclosure failure, see Movie 8. (D) Phalloidin staining of a wild-type embryo at approximately the three-fold stage of elongation. Circumferential filament bundles (CFBs) are clearly visible; the bright staining is one of the four muscle quadrants. (E) A hmp-1(zu278) homozygote stained with phalloidin. CFBs have detached from cell-cell junctions (arrows). (F) A vab-9(e2016) embryo. CFBs aggregate into abnormally large bundles (arrow). Images in (A, B, D, E) courtesy of M. Costa; (C) courtesy of W. B. Raich, from (Costa et al., 1998); used by permission of Rockefeller Press. (F) is courtesy of J. Simske, from (Simske et al., 2003); used by permission of Nature Publishing Group. Scale bar = 5 μm.
C. elegans embryonic morphogenesis comprises a rich array of developmental and cell biological processes that are becoming increasingly tractable. Many of the processes of embryonic morphogenesis exemplify morphogenetic processes found in other organisms (epiboly, cell intercalation, constriction of the apical cytoskeleton). Almost all the molecular pathways so far defined are shared with other organisms, making C. elegans morphogenesis a model for many cellular and developmental phenomena.
As morphogenesis by its nature is a dynamic process, timelapse analysis has been essential to form an accurate picture of morphogenetic processes and defects. Advanced microscopy methods that allow dynamic analysis of subcellular structures will increasingly be needed to explore these processes. Computer aided reconstructions of the cellular dynamics of morphogenetic movements are labor intensive, but likely will become increasingly important in understanding the cellular movements in embryos (Heid et al., 2002; Schnabel et al., 1997). A major future goal will be to test roles for specific cells or subcellular structures by laser microsurgery combined with timelapse recording.
Although genetic and RNA interference analysis has provided insights into many of the processes of epidermal morphogenesis, we are clearly still far from a detailed mechanistic understanding of most morphogenetic movements. As morphogenesis is an essential process, very large scale screens for morphogenetic mutants are labor intensive; with the possible exception of the Hmr or Pat phenotypes, forward screens for morphogenetic defects have not been saturated. With the advent of RNAi hypersensitive strain backgrounds, a more comprehensive picture of the genetic requirements for embryonic morphogenesis should be within reach. Finally, an emerging theme is that many interesting pathways in morphogenesis display a high degree of redundancy: roles of such pathways can only be revealed by quantitative timelapse studies or by use of sensitized genetic backgrounds.
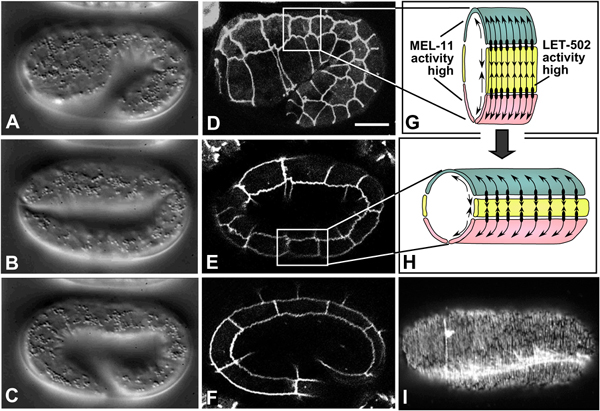
Figure 7. Embryonic elongation. Nomarski images (A-C, left column; lateral view; see also Movie 9) and corresponding DLG-1::GFP images, highlighting epidermal cell boundaries (middle column; D-F), of embryonic elongation. (G, H) Schematic representations of a section of the trunk epidermis corresponding to the boxed regions in panels D and E, respectively. (G, H) are adapted from (Chin-Sang and Chisholm, 2000), with permission of Elsevier Publishing. Ventral cells are shown in pink, seam cells in yellow, and dorsal cells in teal. Contractile forces are produced predominantly by seam epidermal cells (arrows pointing toward one another), and transmitted to dorsal and ventral cells via adherens junctions (black ovals), which anchor circumferential actin filaments bundles (CFBs) in dorsal and ventral cells (black lines and arrows within dorsal and ventral cells). CFBs are thought to transmit and distribute the forces of contraction evenly throughout the epidermis. In seam cells, the activity of LET-502/Rho kinase is high, leading to myosin-dependent contraction; conversely, MEL-11/myosin phosphatase activity is low in seam cells and presumed high in non-seam cells. (I) Phalloidin staining of a wild-type embryo at approximately the two-fold stage of elongation (dorsal view). CFBs are clearly visible. The bright staining is one of the four muscle quadrants (image courtesy of E. William-Masson). Scale bar = 5 μm.
We thank the co-workers mentioned in the Figure and Movie legends for images and 4D movies. Work on C. elegans morphogenesis in our laboratories is supported by NIH grants GM54657 (A.D.C.) and GM58038 (J.H.) and NSF grant IBN0112803 (J.H.).
Andachi, Y. (2004). Caenorhabditis elegans T-box genes tbx-9 and tbx-8 are required for formation of hypodermis and body-wall muscle in embryogenesis. Genes Cells 9, 331–344. Abstract Article
Antoshechkin, I., and Han, M. (2002). The C. elegans evl-20 gene is a homolog of the small GTPase ARL2 and regulates cytoskeleton dynamics during cytokinesis and morphogenesis. Dev. Cell 2, 579–591. Abstract Article
Aspock, G., and Burglin, T.R. (2001). The Caenorhabditis elegans distal-less ortholog ceh-43 is required for development of the anterior hypodermis. Dev. Dyn 222, 403–409. Abstract Article
Bosher, J.M., Hahn, B.S., Legouis, R., Sookhareea, S., Weimer, R.M., Gansmuller, A., Chisholm, A.D., Rose, A.M., Bessereau, J.L., and Labouesse, M. (2003). The Caenorhabditis elegans vab-10 spectraplakin isoforms protect the epidermis against internal and external forces. J. Cell Biol. 161, 757–768. Abstract Article
Brunschwig, K., Wittmann, C., Schnabel, R., Burglin, T.R., Tobler, H., and Muller, F. (1999). Anterior organization of the Caenorhabditis elegans embryo by the labial-like Hox gene ceh-13. Development 126, 1537–1546. Abstract
Chen, W., Chen, S., Yap, S.F., and Lim, L. (1996). The Caenorhabditis elegans p21-activated kinase (CePAK) colocalizes with CeRac1 and CDC42Ce at hypodermal cell boundaries during embryo elongation. J. Biol. Chem. 271, 26362–26368. Abstract Article
Chin-Sang, I.D., and Chisholm, A.D. (2000). Form of the worm: genetics of epidermal morphogenesis in C. elegans. Trends Genet. 16, 544–551. Abstract Article
Chin-Sang, I.D., George, S.E., Ding, M., Moseley, S.L., Lynch, A.S., and Chisholm, A.D. (1999). The ephrin VAB-2/EFN-1 functions in neuronal signaling to regulate epidermal morphogenesis in C. elegans. Cell 99, 781–790. Abstract Article
Chin-Sang, I.D., Moseley, S.L., Ding, M., Harrington, R.J., George, S.E., and Chisholm, A.D. (2002). The divergent C. elegans ephrin EFN-4 functions in embryonic morphogenesis in a pathway independent of the VAB-1 Eph receptor. Development 129, 5499–5510. Abstract Article
Costa, M., Draper, B.W., and Priess, J.R. (1997). The role of actin filaments in patterning the Caenorhabditis elegans cuticle. Dev. Biol. 184, 373–384. Abstract Article
Costa, M., Raich, W., Agbunag, C., Leung, B., Hardin, J., and Priess, J.R. (1998). A putative catenin-cadherin system mediates morphogenesis of the Caenorhabditis elegans embryo. J. Cell Biol. 141, 297–308. Abstract Article
Ding, M., Goncharov, A., Jin, Y., and Chisholm, A.D. (2003). C. elegans ankyrin repeat protein VAB-19 is a component of epidermal attachment structures and is essential for epidermal morphogenesis. Development 130, 5791–5801. Abstract Article
Ding, M., Woo, W.M., and Chisholm, A.D. (2004). The cytoskeleton and epidermal morphogenesis in C. elegans. Exp. Cell Res. 301, 84–90. Abstract Article
Elul, T., and Keller, R. (2000). Monopolar protrusive activity: a new morphogenic cell behavior in the neural plate dependent on vertical interactions with the mesoderm in Xenopus. Dev. Biol. 224, 3–19. Abstract Article
Firestein, B.L., and Rongo, C. (2001). DLG-1 is a MAGUK similar to SAP97 and is required for adherens junction formation. Mol. Biol. Cell 12, 3465–3475. Abstract
Francis, R., and Waterston, R.H. (1991). Muscle cell attachment in Caenorhabditis elegans. J. Cell Biol. 114, 465–479. Abstract Article
Gendreau, S.B., Moskowitz, I.P., Terns, R.M., and Rothman, J.H. (1994). The potential to differentiate epidermis is unequally distributed in the AB lineage during early embryonic development in C. elegans. Dev Biol. 166(2), 770–781. Abstract Article
George, S.E., Simokat, K., Hardin, J., and Chisholm, A.D. (1998). The VAB-1 Eph receptor tyrosine kinase functions in neural and epithelial morphogenesis in C. elegans. Cell. 92(5), 633–643. Abstract Article
Gilleard, J.S., and McGhee, J.D. (2001). Activation of hypodermal differentiation in the Caenorhabditis elegans embryo by GATA transcription factors ELT-1 and ELT-3. Mol. Cell Biol. 21, 2533–2544. Abstract Article
Gilleard, J.S., Shafi, Y., Barry, J.D., and McGhee, J.D. (1999). ELT-3: A Caenorhabditis elegans GATA factor expressed in the embryonic epidermis during morphogenesis. Dev. Biol. 208, 265–280. Abstract Article
Ginzburg, V.E., Roy, P.J., and Culotti, J.G. (2002). Semaphorin 1a and semaphorin 1b are required for correct epidermal cell positioning and adhesion during morphogenesis in C. elegans. Development 129, 2065–2078. Abstract
Gupta, M.C., Graham, P.L., and Kramer, J.M. (1997). Characterization of alpha1(IV) collagen mutations in Caenorhabditis elegans and the effects of alpha1 and alpha2(IV) mutations on type IV collagen distribution. J. Cell Biol. 137, 1185–1196. Abstract Article
Hammarlund, M., Davis, W.S., and Jorgensen, E.M. (2000). Mutations in beta-spectrin disrupt axon outgrowth and sarcomere structure. J. Cell Biol. 149, 931–942. Abstract Article
Hardin, J., and Walston, T. (2004). Models of morphogenesis: the mechanisms and mechanics of cell rearrangement. Curr. Opin. Genet. Dev. 14, 399–406. Abstract Article
Harrington, R.J., Gutch, M.J., Hengartner, M.O., Tonks, N.K., and Chisholm, A.D. (2002). The C. elegans LAR-like receptor tyrosine phosphatase PTP-3 and the VAB-1 Eph receptor tyrosine kinase have partly redundant functions in morphogenesis. Development 129, 2141–2153. Abstract
Heid, P.J., Raich, W.B., Smith, R., Mohler, W.A., Simokat, K., Gendreau, S.B., Rothman, J.H., and Hardin, J. (2001). The zinc finger protein DIE-1 is required for late events during epithelial cell rearrangement in C. elegans. Dev. Biol. 236, 165–180. Abstract Article
Heid, P.J., Voss, E., and Soll, D.R. (2002). 3D-DIASemb: a computer-assisted system for reconstructing and motion analyzing in 4D every cell and nucleus in a developing embryo. Dev. Biol. 245, 329–347. Abstract Article
Hresko, M.C., Schriefer, L.A., Shrimankar, P., and Waterston, R.H. (1999). Myotactin, a novel hypodermal protein involved in muscle-cell adhesion in Caenorhabditis elegans. J. Cell Biol. 146, 659–672. Abstract Article
Hresko, M.C., Williams, B.D., and Waterston, R.H. (1994). Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J. Cell Biol. 124, 491–506. Abstract Article
Huang, C.C., Hall, D.H., Hedgecock, E.M., Kao, G., Karantza, V., Vogel, B.E., Hutter, H., Chisholm, A.D., Yurchenco, P.D., and Wadsworth, W.G. (2003). Laminin α subunits and their role in C. elegans development. Development 130, 3343–3358. Abstract Article
Jacinto, A., Woolner, S., and Martin, P. (2002). Dynamic analysis of dorsal closure in Drosophila: from genetics to cell biology. Dev. Cell 3, 9–19. Abstract Article
Junkersdorf, B., and Scheierenberg, E. (1992). Embryogenesis in C. elegans after elimination of individual blastomeres or induced alteration of the cell-division order. Roux's Arch. Dev. Biol. 202, 17–22. Article
Koppen, M., Simske, J.S., Sims, P.A., Firestein, B.L., Hall, D.H., Radice, A.D., Rongo, C., and Hardin, J.D. (2001). Cooperative regulation of AJM-1 controls junctional integrity in Caenorhabditis elegans epithelia. Nat. Cell Biol. 3, 983–991. Abstract Article
Labouesse, M., Sookhareea, S., and Horvitz, H.R. (1994). The Caenorhabditis elegans gene lin-26 is required to specify the fates of hypodermal cells and encodes a presumptive zinc-finger transcription factor. Development 120, 2359–2368. Abstract
Lundquist, E.A., Reddien, P.W., Hartwieg, E., Horvitz, H.R., and Bargmann, C.I. (2001). Three C. elegans Rac proteins and several alternative Rac regulators control axon guidance, cell migration and apoptotic cell phagocytosis. Development 128, 4475–4488. Abstract
Mango, S.E., Lambie, E.J., and Kimble, J. (1994). The pha-4 gene is required to generate the pharyngeal primordium of Caenorhabditis elegans. Development 120, 3019–3031. Abstract
McKeown, C., Praitis, V., and Austin, J. (1998). sma-1 encodes a betaH-spectrin homolog required for Caenorhabditis elegans morphogenesis. Development 125, 2087–2098. Abstract
McMahon, L., Legouis, R., Vonesch, J.L., and Labouesse, M. (2001). Assembly of C. elegans apical junctions involves positioning and compaction by LET-413 and protein aggregation by the MAGUK protein DLG-1. In J. Cell Sci. 114, 2265–2277. Abstract
Mohler, W.A., Shemer, G., del Campo, J.J., Valansi, C., Opoku-Serebuoh, E., Scranton, V., Assaf, N., White, J.G., and Podbilewicz, B. (2002). The type I membrane protein EFF-1 is essential for developmental cell fusion. Dev Cell. 2(3), 355–362. Abstract Article
Moorthy, S., Chen, L., and Bennett, V. (2000). Caenorhabditis elegans beta-G spectrin is dispensable for establishment of epithelial polarity, but essential for muscular and neuronal function. J. Cell Biol. 149, 915–930. Abstract Article
Munro, E.M., and Odell, G.M. (2002). Polarized basolateral cell motility underlies invagination and convergent extension of the ascidian notochord. Development 129, 13–24. Abstract
Norman, K.R., and Moerman, D.G. (2002). Alpha spectrin is essential for morphogenesis and body wall muscle formation in Caenorhabditis elegans. J. Cell Biol. 157, 665–677. Abstract Article
Novelli, J., Ahmed, S., and Hodgkin, J. (2004). Gene interactions in Caenorhabditis elegans define DPY-31 as a candidate procollagen C-proteinase and SQT-3/ROL-4 as its predicted major target. Genetics 168, 1259–1273. Abstract Article
Page, B.D., Zhang, W., Steward, K., Blumenthal, T., and Priess, J.R. (1997). ELT-1, a GATA-like transcription factor, is required for epidermal cell fates in Caenorhabditis elegans embryos. Genes Dev. 11(13), 1651–1661. Abstract
Pasterkamp, R.J., and Kolodkin, A.L. (2003). Semaphorin junction: making tracks toward neural connectivity. Curr. Opin. Neurobiol. 13, 79–89. Abstract Article
Pettitt, J., Cox, E.A., Broadbent, I.D., Flett, A., and Hardin, J. (2003). The Caenorhabditis elegans p120 catenin homologue, JAC-1, modulates cadherin-catenin function during epidermal morphogenesis. J. Cell Biol. 162, 15–22. Abstract Article
Piekny, A.J., Johnson, J.L., Cham, G.D., and Mains, P.E. (2003). The Caenorhabditis elegans nonmuscle myosin genes nmy-1 and nmy-2 function as redundant components of the let-502/ρ-binding kinase and mel-11/myosin phosphatase pathway during embryonic morphogenesis. Development 130, 5695–5704. Abstract Article
Piekny, A.J., Wissmann, A., and Mains, P.E. (2000). Embryonic morphogenesis in Caenorhabditis elegans integrates the activity of LET-502 Rho-binding kinase, MEL-11 myosin phosphatase, DAF-2 insulin receptor and FEM-2 PP2c phosphatase. Genetics 156, 1671–1689. Abstract
Pocock, R., Ahringer, J., Mitsch, M., Maxwell, S., and Woollard, A. (2004). A regulatory network of T-box genes and the even-skipped homologue vab-7 controls patterning and morphogenesis in C. elegans. Development 131, 2373–2385. Abstract Article
Praitis, V., Ciccone, E., and Austin, J. (2005). SMA-1 spectrin has essential roles in epithelial cell sheet morphogenesis in C. elegans. Dev. Biol. 283(1), 157–170. Abstract Article
Priess, J.R., and Hirsh, D.I. (1986). Caenorhabditis elegans morphogenesis: the role of the cytoskeleton in elongation of the embryo. Dev. Biol. 117, 156–173. Abstract Article
Quintin, S., Michaux, G., McMahon, L., Gansmuller, A., and Labouesse, M. (2001). The Caenorhabditis elegans gene lin-26 can trigger epithelial differentiation without conferring tissue specificity. Dev. Biol. 235, 410–421. Abstract Article
Raich, W.B., Agbunag, C., and Hardin, J. (1999). Rapid epithelial-sheet sealing in the Caenorhabditis elegans embryo requires cadherin-dependent filopodial priming. Curr. Biol. 9, 1139–1146. Abstract Article
Roberts, B., Clucas, C., and Johnstone, I.L. (2003). Loss of SEC-23 in Caenorhabditis elegans causes defects in oogenesis, morphogenesis, and extracellular matrix secretion. Mol. Biol. Cell 14, 4414–4426. Abstract Article
Roy, P.J., Zheng, H., Warren, C.E., and Culotti, J.G. (2000). mab-20 encodes Semaphorin-2a and is required to prevent ectopic cell contacts during epidermal morphogenesis in Caenorhabditis elegans. Development 127, 755–767. Abstract
Rugarli, E.I., Di Schiavi, E., Hilliard, M.A., Arbucci, S., Ghezzi, C., Facciolli, A., Coppola, G., Ballabio, A., and Bazzicalupo, P. (2002). The Kallmann syndrome gene homolog in C. elegans is involved in epidermal morphogenesis and neurite branching. Development 129, 1283–1294. Abstract
Sawa, M., Suetsugu, S., Sugimoto, A., Miki, H., Yamamoto, M., and Takenawa, T. (2003). Essential role of the C. elegans Arp2/3 complex in cell migration during ventral enclosure. J. Cell Sci. 116, 1505–1518. Abstract Article
Schnabel, R., Hutter, H., Moerman, D., and Schnabel, H. (1997). Assessing normal embryogenesis in Caenorhabditis elegans using a 4D microscope: variability of development and regional specification. Dev. Biol. 184, 234–265. Abstract Article
Severson, A.F., Baillie, D.L., and Bowerman, B. (2002). A Formin Homology protein and a profilin are required for cytokinesis and Arp2/3-independent assembly of cortical microfilaments in C. elegans. Curr. Biol. 12, 2066–2075. Abstract Article
Shelton, C.A., Carter, J.C., Ellis, G.C., and Bowerman, B. (1999). The nonmuscle myosin regulatory light chain gene mlc-4 is required for cytokinesis, anterior-posterior polarity, and body morphology during Caenorhabditis elegans embryogenesis. J. Cell Biol. 146, 439–451. Abstract Article
Simske, J.S., Koppen, M., Sims, P., Hodgkin, J., Yonkof, A., and Hardin, J. (2003). The cell junction protein VAB-9 regulates adhesion and epidermal morphology in C. elegans. Nat. Cell Biol. 5, 619–625. Abstract Article
Soto, M.C., Qadota, H., Kasuya, K., Inoue, M., Tsuboi, D., Mello, C.C., and Kaibuchi, K. (2002). The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev. 16, 620–632. Abstract Article
Starr, D.A., Hermann, G.J., Malone, C.J., Fixsen, W., Priess, J.R., Horvitz, H.R., and Han, M. (2001). unc-83 encodes a novel component of the nuclear envelope and is essential for proper nuclear migration. Development 128, 5039–5050. Abstract
Sulston, J.E., Schierenberg, E., White, J.G., and Thomson, J.N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119. Abstract Article
Thomas-Virnig, C.L., Sims, P.A., Simske, J.S., and Hardin, J. (2004). The inositol 1,4,5-trisphosphate receptor regulates epidermal cell migration in Caenorhabditis elegans. Curr. Biol. 14, 1882–1887. Abstract Article
Van Auken, K., Weaver, D., Robertson, B., Sundaram, M., Saldi, T., Edgar, L., Elling, U., Lee, M., Boese, Q., and Wood, W.B. (2002). Roles of the Homothorax/Meis/Prep homolog UNC-62 and the Exd/Pbx homologs CEH-20 and CEH-40 in C. elegans embryogenesis. Development 129, 5255–5268. Abstract
Wang, X., Roy, P.J., Holland, S.J., Zhang, L.W., Culotti, J.G., and Pawson, T. (1999). Multiple ephrins control cell organization in C. elegans using kinase-dependent and -independent functions of the VAB-1 Eph receptor. Mol. Cell 4, 903–913. Abstract Article
Williams, B.D., and Waterston, R.H. (1994). Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J. Cell Biol. 124, 475–490. Abstract Article
Williams-Masson, E.M., Heid, P.J., Lavin, C.A., and Hardin, J. (1998). The cellular mechanism of epithelial rearrangement during morphogenesis of the Caenorhabditis elegans dorsal hypodermis. Dev. Biol. 204, 263–276. Abstract Article
Williams-Masson, E.M., Malik, A.N., and Hardin, J. (1997). An actin-mediated two-step mechanism is required for ventral enclosure of the C. elegans hypodermis. Development 124, 2889–2901. Abstract
Wissmann, A., Ingles, J., and Mains, P.E. (1999). The Caenorhabditis elegans mel-11 myosin phosphatase regulatory subunit affects tissue contraction in the somatic gonad and the embryonic epidermis and genetically interacts with the Rac signaling pathway. Dev. Biol. 209, 111–127. Abstract Article
Wissmann, A., Ingles, J., McGhee, J.D., and Mains, P.E. (1997). Caenorhabditis elegans LET-502 is related to ρ-binding kinases and human myotonic dystrophy kinase and interacts genetically with a homolog of the regulatory subunit of smooth muscle myosin phosphatase to affect cell shape. Genes Dev. 11, 409–422. Abstract
*Edited by James R. Priess and Geraldine Seydoux. Last revised May 19, 2005 . Published December 01, 2005. This chapter should be cited as: Chisholm, A.D. and Hardin, J. Epidermal morphogenesis (December 01, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.35.1 , http://www.wormbook.org.
§To whom correspondence should be addressed. E-mail: jdhardin@wisc.edu
 All journal content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All journal content, except where otherwise noted, is licensed under a Creative Commons Attribution License.