Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Cell-division control affects many aspects of development. Caenorhabditis elegans cell-cycle genes have been identified over the past decade, including at least two distinct Cyclin-Dependent Kinases (CDKs), their cyclin partners, positive and negative regulators, and downstream targets. The balance between CDK activation and inactivation determines whether cells proceed through G1 into S phase, and from G2 to M, through regulatory mechanisms that are conserved in more complex eukaryotes. The challenge is to expand our understanding of the basic cell cycle into a comprehensive regulatory network that incorporates environmental factors and coordinates cell division with growth, differentiation and tissue formation during development. Results from several studies indicate a critical role for CKI-1, a CDK inhibitor of the Cip/Kip family, in the temporal control of cell division, potentially acting downstream of heterochronic genes and dauer regulatory pathways.
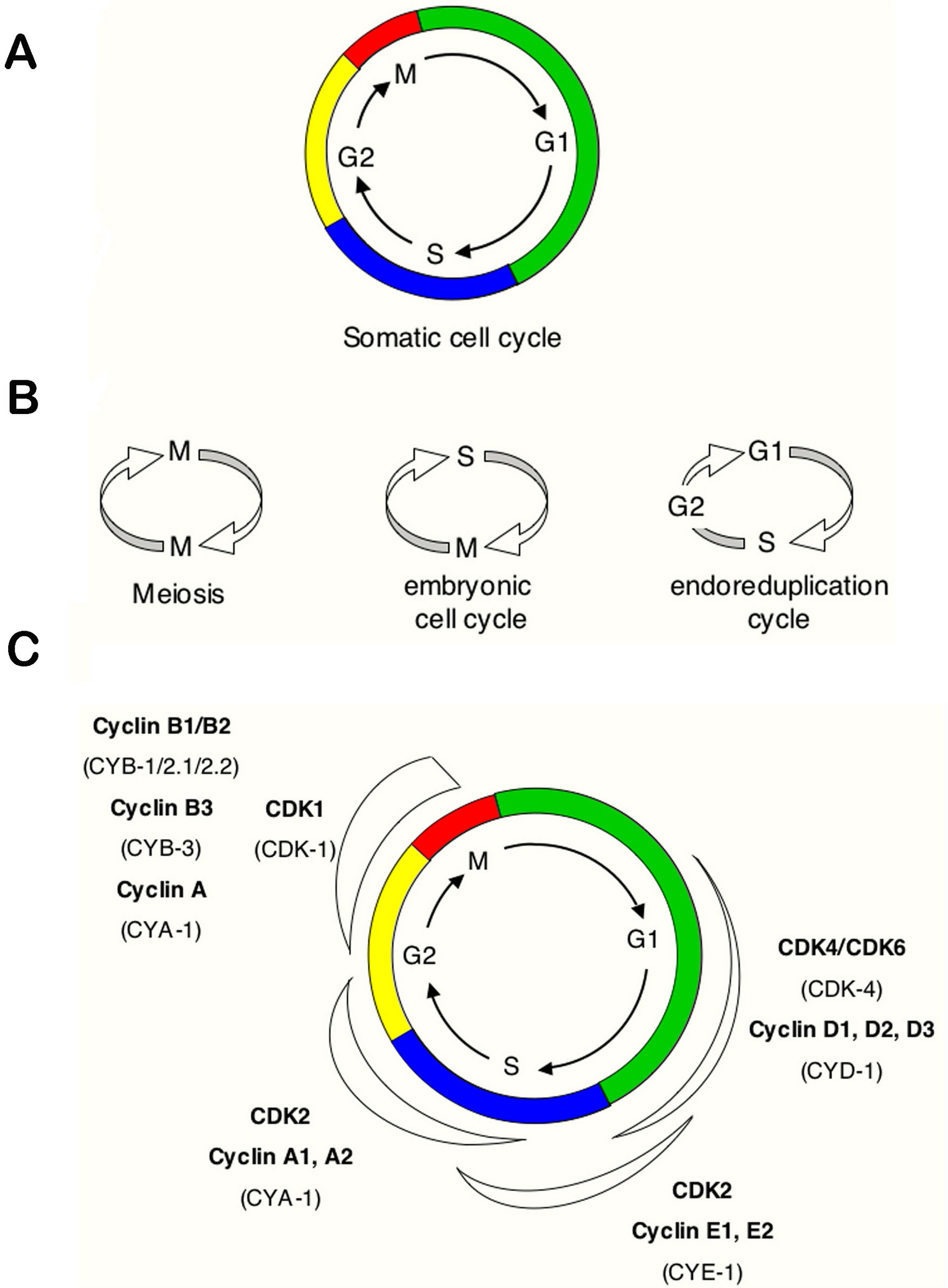
Animal development from a single-cell zygote to fertile adult requires many rounds of cell division. During each division, cells complete an ordered series of events that collectively form the "cell cycle". This cycle includes accurate duplication of the genome during the DNA synthesis phase (S phase), and segregation of complete sets of chromosomes to each of the daughter cells in M phase (Figure 1A). The somatic cell cycle also contains "Gap" phases, known as G1, which connects the completion of M phase to initiation of S phase in the next cycle, and G2, which separates the S and M phases. Dependent on environmental and developmental signals, cells in G1 may temporarily or permanently leave the cell cycle and enter a quiescent or arrested phase known as G0.
|
Figure 1. Simple representations of the cell cycle. (A) a typical (somatic) cell cycle, which can be divided in four sequential phases: G1, S, G2 and M. M phase consist of nuclear division (mitosis) and cytoplasmic division (cytokinesis) (B) variant cell cycles in which specific phases are omitted. (C) approximate time of activity for different combinations of cyclins and CDKs, based on studies of mammalian cyclins and CDKs. C. elegans family members are indicated between brackets. Shapes outside the cycle indicate increase and reduction of corresponding CDK/cyclin activity.
During development, variations of this typical somatic division cycle are used to fulfill specific requirements (Figure 1B). These include rapid embryonic cell cycles that lack G1 and G2 phases, meiotic cell cycles that allow formation of haploid gametes, and endoreduplication (or: "endoreplication") cycles in which S phases are not followed by mitosis. These variant cell cycles form part of the stereotypical pattern of cell divisions during C. elegans development.
Cell external signals and cell intrinsic information together determine whether cells enter a division cycle. In general, external signals affect this decision only until cells commit to go through the entire cycle, at a time in G1 known as "START" in yeast and "Restriction point" in mammals. From there on, progression through the cell cycle is controlled intrinsically by the cell-cycle machinery. The basic components of this machinery are conserved in all eukaryotes. Consequently, findings based on genetics in yeast, biochemistry in frog eggs and tissue culture of mammalian cells have all come together and generated a substantial molecular understanding of cell-cycle regulation.
The next level of questions include: how is cell division temporally and spatially controlled during development, and how is progression through the cycle coordinated with cell growth, differentiation, migration, and death? C. elegans is well suited to study developmental control of cell division and to address how nutritional signals, differentiation factors, checkpoint controls, heterochronic genes, and dauer regulatory pathways act upon the cell-cycle machinery to affect cell-cycle entry and exit. Insights into the basic regulators of cell-cycle progression in C. elegans form the foundation for such studies and are the focus of this chapter.
The collective results from studies in various eukaryotes have demonstrated that progression through the cell-division cycle is driven by activation and inactivation of cyclin-dependent kinases (CDKs), which trigger the transition to subsequent phases of the cycle. CDKs are small serine/threonine protein kinases that require association with a cyclin subunit for their activation. In yeast, a single CDK (p34CDC28 in Saccharomyces cerevisiae and p34cdc2 in Schizosaccharomyces pombe) acts with different cyclins to promote progression through G1, S and G2/M. Metazoans, including C. elegans, use not only a variety of cyclins but also multiple catalytic subunits (Figure 1C).
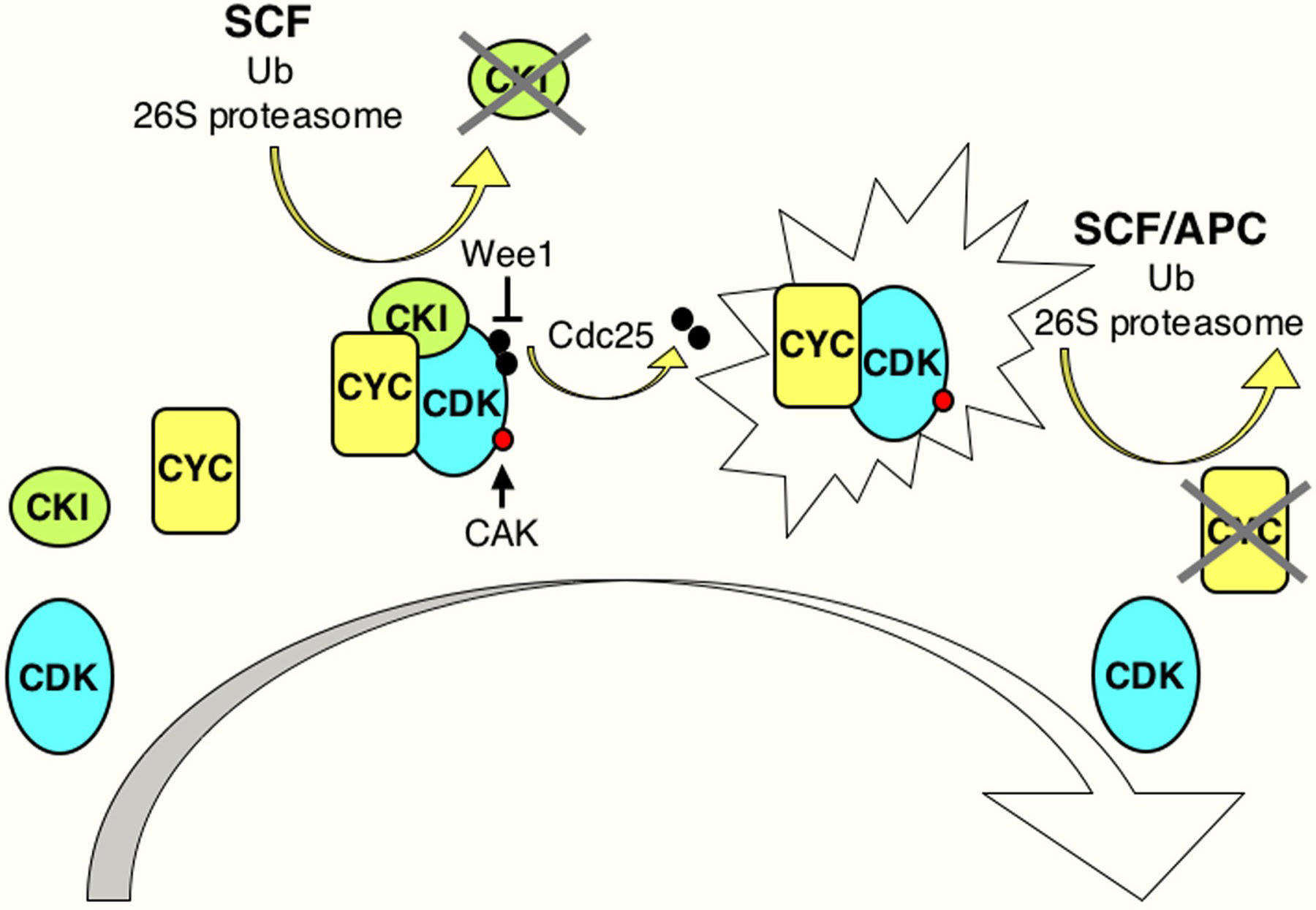
Many levels of regulation impinge upon the CDKs to impose tight control over cell-cycle progression. Such regulation involves controlled expression and destruction of cyclins, activating and inhibitory phosphorylation and dephosphorylation of the CDKs, and expression and destruction of inhibitory proteins that associate with CDKs, or CDK/cyclin complexes (Figure 2).
|
Figure 2. Model illustrating general aspects of CDK regulation. CDK activation requires cyclin (CYC) expression and association. Cyclin/CDK complexes are kept inactive through association with CDK-inhibitory proteins (CKIs) and inhibitory phosphorylation by Wee1/Myt1 kinases (black circles). Activation requires ubiquitin-dependent proteolysis of the CKI, phosphorylation of the CDK by a CDK-activating kinase (CAK; red circle), and removal of the inhibitory phosphates by a Cdc25 phosphatase. Cyclin destruction leads to inactivation. Ubiquitin-dependent proteolysis of cell cycle regulators in late G1 and S involves cullin-based E3 ligases such as SCF, while in M phase and early G1 the anaphase-promoting complex (APC) is active. The exclamation figure denotes the active kinase complex, the large arrow indicates time.
The paradigm for cell-cycle regulation through activation and inactivation of CDKs applies to all eukaryotes. However, differences do exist: certain control elements, such as the CDK inhibitory proteins (CKIs), show little resemblance between yeast and mammals. In addition, some regulators are absent in single cell eukaryotes, including the pRb and E2F families, and nearly all regulatory genes have expanded into subfamilies with multiple members in mammals. Studies over the last decade have shown that cell-cycle control in C. elegans uses well-recognizable homologs of nearly all mammalian regulators, often represented by just a single member (Table 1). As an exception to the rule, the pINK family of Cdk4/6 kinase inhibitors has not as yet been identified in C. elegans. Genetic studies have placed the C. elegans cell-cycle genes into pathways that resemble those in mammals, and novel regulatory elements have been discovered.
Table 1. C. elegans cell-cycle mutants (for references: see text)
| Gene name | Alternate | Homolog | Presumed function | Cell-cycle phenotype (loss of function) |
|---|---|---|---|---|
| Cyclin-dependent kinases | ||||
| cdk-1 | ncc-1 | Cdk1 | Promotes M phase entry/progression | G2 arrest, starting in L1. RNAi: one-cell arrest |
| cdk-4 | Cdk4/Cdk6 | Promotes Progression through G1 | G1 arrest, starting late embryogenesis | |
| cdk-7 | Cdk7 | CDK activating kinase/Pol II CTD kinase | cdk-7(ax224 RNAi): one-cell arrest | |
| Cyclins | ||||
| cyd-1 | Cyclin D1/D2/D3 | G1 Cyclin, promotes progression through G1 | G1 arrest, starting late in embryogenesis | |
| cye-1 | evl-10 | Cyclin E1/E2 | G1/S Cyclin | Late larval defects. (RNAi: 100-cell arrest) |
| Inhibitors | ||||
| cki-1 | Kip1 | Cip/Kip family CDK inhibitor. Inhibits G1/S transition. | Extra cell division (failure to arrest) | |
| CDK inhibitory phosphorylation | ||||
| cdc-25.1 | Cdc25 | CDK activating dual specificity phosphatase | Sterile lavae (RNAi; early embryonic defects) | |
| wee-1.3 | spe-37 | Myt1/Wee1 | CDK inhibitory kinase | Embryonic and larval lethal |
| Rb/E2F related | ||||
| lin-35 | pRb/p107 | Co-repressor. Negative regulator G1 | Suppresses cdk-4/cyd-1, synthetic extra division | |
| efl-1 | mex-2 | E2F4/5 | Transcription factor. Negative regulator G1 | Similar to lin-35 loss of function (but weaker) |
| dpl-1 | lin-55, mex-4 | DP1 | Transcription factor. Pos/Neg regulator G1 | Reduced cell division, but suppresses cdk-4/cyd-1 |
| lin-36 | unknown | Zn2+ finger protein. | Similar to lin-35 loss of function | |
| lin-9 | Mip130/TWIT | Component pRb repressor complex | Weak suppression cyd-1/cdk-4 | |
| lin-15B | unknown | Synthetic extra division, suppresses cdk-4/cyd-1 | ||
| SCF | ||||
| cul-1 | lin-19 | Cul1 | Cullin subunit SCF. G1 cyclin degradation | Hyperplasia, resulting from failure in cell-cycle exit |
| lin-23 | F box factor | SCF substrate specificity factor | Hyperplasia, resulting from failure in cell-cycle exit | |
| APC | ||||
| mat-1 | pod-5 | APC3, Cdc27 | Component APC E3 Ubiquitin ligase | Arrest at metaphase-to-anaphase transition meiosis |
| mat-2 | pod-3, evl-22 | APC1 | Component APC E3 Ubiquitin ligase | Arrest at metaphase-to-anaphase transition meiosis |
| mat-3 | pod-4 | APC8, Cdc23 | Component APC E3 Ubiquitin ligase | Arrest at metaphase-to-anaphase transition meiosis |
| emb-27 | pod-6 | APC6, Cdc16 | Component APC E3 Ubiquitin ligase | Arrest at metaphase-to-anaphase transition meiosis |
| emb-30 | APC4 | Component APC E3 Ubiquitin ligase | Arrest at metaphase-to-anaphase transition | |
| DNA damage/DNA replication checkpoint | ||||
| mrt-2 | Rad1 | DNA-damage checkpoint protein | Deficient in apoptosis in response to DNA damage | |
| clk-2 | rad-5 | ScTel2p | DNA-damage checkpoint protein | Deficient in apoptosis in response to DNA damage |
| hus-1 | Hus1 | DNA-damage checkpoint protein | Deficient in apoptosis in response to DNA damage | |
| cep-1 | p53 | Transcription factor DNA-damage checkpoint | Deficient in apoptosis in response to DNA damage | |
| Spindle assembly checkpoint | ||||
| mdf-1 | MAD1 | Spindle assembly checkpoint protein | Mitotic arrest defective in nocodazole. Lethal, sterile | |
| san-1 | MAD3 | Mitotic checkpoint kinase | Failure to arrest in metaphase in anoxic conditions | |
The C. elegans genome encodes multiple members of the cyclin-dependent kinase (CDK) family. At least two CDKs, CDK-1 and CDK-4, are essential for cell-cycle progression (Boxem et al., 1999; Boxem and van den Heuvel, 2001; Park and Krause, 1999). These CDKs act at distinct times in the cell cycle and use specific cyclin partners, similar to their mammalian orthologs (Table 1).
CDK-1, previously known as NCC-1 for nematode cell cycle, was identified based on its close similarity to the prototypical yeast CDK (Mori et al., 1994). In contrast to yeast, but similar to mammalian Cdk1, cdk-1/(ncc-1) is specifically required for G2/M progression and not for G1 or S phase (Boxem et al., 1999). Maternal cdk-1 product suffices for embryogenesis, and candidate null mutant animals arrest cell division during L1 development. Several observations indicate that the post-embryonic precursor cells in these mutants arrest in G2 phase: such cells show normal expression of the rnr::GFP reporter and BrdU incorporation during S phase, but fail to proceed into mitosis (as indicated by absence of chromosome condensation and nuclear envelope breakdown). Moreover, endoreduplication cycles, which skip M phase, continue in cdk-1 mutants and intestinal nuclei accumulate the normal 32n DNA content. Following RNAi of cdk-1 in adult hermaphrodites, oocytes show delayed meiotic maturation, form an eggshell upon fertilization, but neither align nor segregate homologous chromosomes. Thus, cdk-1 is required for meiotic as well as mitotic M phase.
CDK-1 likely acts with mitotic cyclins of the A and B subfamilies (Kreutzer et al., 1995). A single full-length cyclin A gene (cya-1) is expressed in C. elegans, as well as three typical B-type cyclins (cyb-1, cyb-2.1 and cyb-2.2), and a distinct member of the cyclin B3 subfamily (cyb-3). While cyb-1 and cyb-3 each are individually required during embryonic development, simultaneous inactivation of these mitotic cyclins causes a more severe phenotype: cyb-1;cyb-3(RNAi) embryos arrest at the one-cell stage and resemble cdk-1(RNAi) embryos (our unpublished results). These data support the notion that different mitotic cyclins have functions that are partly distinct and partly overlapping.
The cdk-4 Cdk4/6 kinase and cyd-1 D-type cyclin genes are required for progression through G1 phase during larval development (Boxem and van den Heuvel, 2001; Park and Krause, 1999). CYD-1 and CDK-4 likely act in complex, as indicated by their direct interaction in vitro and close similarity in null phenotypes (Park and Krause, 1999). In contrast to larval divisions, only a few very late embryonic divisions depend on cyd-1/cdk-4 activity (Boxem and van den Heuvel, 2001; Yanowitz and Fire, 2005). Possible explanations for this difference include that the early embryonic divisions lack a G1 phase and therefore will not need a G1 cyclin or CDK. Also, as one of its most important functions, cyd-1 and cdk-4 act to antagonize the transcriptional repressor lin-35 Rb (see below; (Boxem and van den Heuvel, 2001). In the absence of cyd-1/cdk-4 function, lin-35 Rb may inappropriately repress cell-cycle genes, but this cannot prevent divisions that are driven by maternal products. Also, cyd-1 and cdk-4 could primarily promote growth, as in Drosophila (Datar et al., 2000; Meyer et al., 2000), which is not incorporated in the embryonic divisions. However, larval divisions arrest in G1 while cells continue to grow in the mutants, and growth retardation occurs later (Boxem and van den Heuvel, 2001). Therefore, absence of G1 phases and maternal contribution of DNA replication components likely explain the limited requirement for cyd-1/cdk-4 during embryogenesis
Taken together, CDK-1 and CDK-4 act in G2/M and G1, respectively, like their mammalian orthologs Cdk-1 and Cdk4/6. It is currently not clear whether C. elegans also uses a Cdk2 ortholog, which acts subsequent to Cdk4/6 in mammals to promote G1/S and S phase progression. The best candidate is K03E5.3, which shares 43% amino-acid identity with human Cdk2 (Boxem et al., 1999). Inhibition of this gene by RNAi resulted in a variable phenotype, with animals arresting during embryogenesis, during early or late larval development, and as sterile adults.
In other metazoans, Cdk2 acts with Cyclin E to promote S phase entry. C. elegans cye-1 Cyclin E deletion animals show surprisingly normal development until the L3 stage, at which time the VPC divisions proceed slowly and incompletely (Fay and Han, 2000). This modest phenotype apparently depends on long lasting maternal function, as RNAi results in embryonic lethality at approximately the hundred-cell stage (Brodigan et al., 2003; Fay and Han, 2000).
Several other members of the Cdk superfamily are present in C. elegans, including a Cdk7/Mo15 ortholog (Boxem et al., 1999; Liu and Kipreos, 2000). Cdk7 was identified as CDK-activating kinase (CAK) in mammalian cells, but also as part of the TFIIH transcription factor, responsible for phosphorylating the C-terminal domain (CTD) of RNA polymerase II (Fisher and Morgan, 1994; Shiekhattar et al., 1995). The combination of a temperature-sensitive mutation and RNAi of cdk-7 resulted in one-cell arrest similar to cdk-1(RNAi) embryos (Wallenfang and Seydoux, 2002). In addition, partial inactivation of cdk-7 interfered with transcription and phosphorylation of the RNA polymerase CTD. These data support dual activities of CDK-7 as both CDK-activating kinase (CAK) and CTD kinase in vivo.
Several other CDKs are likely to act independent of the cell cycle. CDK-5 is remarkably close to human Cdk5, sharing 74% identity at the amino-acid level, which has neuronal functions (Cruz and Tsai, 2004). Two other CDKs, CDK-8 and CDK-9, likely are involved specifically in regulating transcription (Liu and Kipreos, 2000; Shim et al., 2002).
Association with small inhibitory proteins is a universal mechanism of CDK regulation (Sherr and Roberts, 1999), though the CKIs (Cyclin-dependent Kinase Inhibitors) involved are highly divergent between yeasts and metazoans. Three different proteins, p21Cip1, p27Kip1 and p57Kip2, form the "CDK inhibitory Protein/Kinase Inhibitor protein" (Cip/Kip) family in mammals. The C. elegans genome encodes two members of this family: CKI-1 and CKI-2 (Feng et al., 1999; Hong et al., 1998). Although both predicted proteins are similarly close in amino-acid sequence to p21Cip1 and p27Kip1, only CKI-1 appears to act generally in cell-cycle control (Boxem and van den Heuvel, 2001; Feng et al., 1999; Fukuyama et al., 2003; Hong et al., 1998).
Several results have shown that CKI-1 acts to promote cell-cycle arrest throughout development, analogous to p27Kip1 in mammals and Dacapo in flies. Absence of cki-1 as a result of the mnDf100 deletion results in embryonic arrest with hyperplasia in multiple lineages, including the intestinal and hypodermal lineages, as well as increased apoptosis and defects in morphogenesis (Fukuyama et al., 2003). mnDf100 also eliminates cki-2 and other genes, but cki-1 genomic sequences partly suppress the phenotype. Moreover, similar defects were observed in a fraction of cki-1(RNAi) embryos. However, RNAi usually causes incomplete inactivation of cki-1 and predominantly gives rise to sterile adults with extra divisions in the postembryonic lineages and extra gonad arms (Shiva phenotype; Hong et al., 1998). Postembryonic precursor cells in such cki-1(RNAi) animals fail to arrest in G1 and ectopically express the S phase marker rnr::gfp. Thus, cki-1 Kip1 function is rate limiting for S phase entry, particularly in cells that enter a prolonged quiescent state before re-entering the cell cycle.
Interestingly, loss of cki-1 Kip1 also affects aspects of cell-fate determination, as the extra distal tip cells (DTCs) in cki-1(RNAi) animals are derived from a different cell type and not from DTC duplication (Kostic et al., 2003).
The tumor suppressor pRb is a well-known transcriptional repressor of, among others, genes involved in S phase progression such as cyclin E. Proteins of the pRb family exert this role through association with transcription factors, primarily with E2F/DP heterodimers (together referred to as "E2F"; Stevaux and Dyson, 2002). This association prevents "activating E2Fs" from promoting transcription. In addition, to inhibit transcription, "repressive E2Fs" recruit pRb family members and associated chromatin remodeling complexes such as the Nucleosome Remodeling and Deacetylase (NuRD) complex.
Several members of the Rb/E2F pathway in C. elegans have been identified as class B synthetic Multivulva (synMuv) genes (see Vulval development). As such, lin-35 Rb, efl-1 E2F4/5, dpl-1 DP and several putative NuRD-complex components all act in a redundant pathway to antagonize Ras-mediated induction of the vulval cell fate. The fact that these genes have similar, rather than opposite, loss-of-function phenotypes provided strong in vivo evidence that E2F/DP can act as a transcriptional repressor, in concert with pRb and NuRD. The contribution of lin-35 Rb in vulval precursor cell (VPC) determination is non cell-autonomous (Myers and Greenwald, 2005), and cannot be explained by lack of cell-cycle control in these cells.
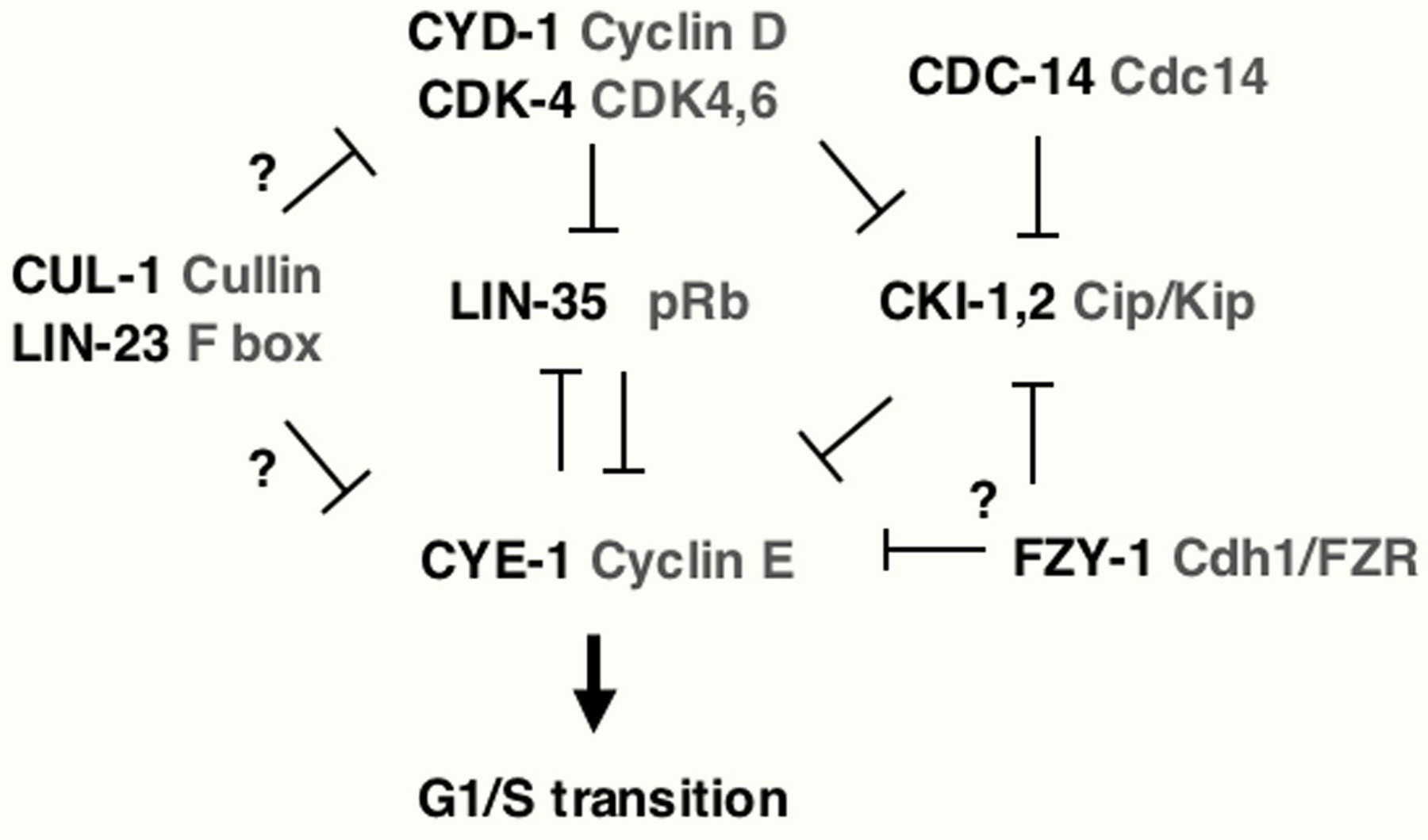
Homozygous lin-35 Rb mutants do not display a prominent increase in cell division, although a fraction of such animals form extra intestinal nuclei (Saito et al., 2004). However, the contribution of lin-35 Rb to negative regulation of G1 progression became apparent in double mutant combinations. Inactivation of lin-35 substantially rescues the larval arrest of cell division in mutants that lack the positive G1 regulators cyd-1 and cdk-4 (Boxem and van den Heuvel, 2001). In addition, lin-35 inactivation synergistically increases the number of extra cell divisions when combined with negative G1 regulators, such as cki-1 Cip/Kip, cdc-14 Cdc14 and fzr-1 Cdh1/FZR (Boxem and van den Heuvel, 2001; Fay et al., 2002; Saito et al., 2004). Together, such results indicate that lin-35 acts redundantly to inhibit G1 progression, likely downstream of cyd-1 and cdk-1 and parallel of cki-1, fzr-1 and cdc-14 (Figure 3).
Examination of additional double mutant combinations revealed that some of the other synMuv class B genes also contribute to G1 control (Boxem and van den Heuvel, 2002; Fay et al., 2002). Specifically, efl-1 E2F negatively regulates cell-cycle entry, while dpl-1 DP appears to act both as a positive and negative regulator. In addition, a negative G1 regulatory function was identified for lin-9 Mip130/TWIT, as well as lin-15B and lin-36, which encode novel proteins. Class A synMuv genes and class B genes that encode NURD components have not been observed to affect the cell cycle.
Candidate targets of Rb/E2F regulation in C. elegans include cye-1 Cyclin E and rnr-1, which encodes the ribonucleotide reductase large subunit. These genes have multiple E2F-binding sites within their promoter regions (Brodigan et al., 2003; Hong et al., 1998) and are regulated by E2F in other species. Several genetic observations are also consistent with lin-35 acting upstream of cye-1 to repress its transcription (Boxem and van den Heuvel, 2001; our unpublished observations).
In all eukaryotes studied, CDKs are regulated by phosphorylation and dephosphorylation of critical residues. Important targets of this regulation are the neighboring threonine and tyrosine in the ATP-binding loop (GXGTYG, corresponding to Thr14 and Tyr15 in fission yeast p34cdc2). Phosphorylation of these residues by the related Wee1 and Myt1 kinases prevents activation of the CDK/cyclin complex. Members of the Cdc25 family of dual-specificity phosphatases counteract Wee1/Myt1 phosphorylation and control the appropriate timing of CDK activation (Figure 2).
Inhibitory phosphorylation allows for developmental regulation of CDK activity, as demonstrated for the String and Twine phosphatases in Drosophila. In C. elegans, two different Wee1/Myt1 kinases are expressed (wee-1.1 and wee-1.3), as are four phosphatases of the Cdc25 family (cdc-25.1 to cdc-25.4). Despite the multiplicity, each family contains at least one essential member: homozygous wee-1.3 null mutants are embryonic lethal and cdc-25.1 null mutants develop into sterile adults (Ashcroft and Golden, 2002; Lamitina and L'Hernault, 2002). In the latter mutants, maternal product likely masks the embryonic function of cdc-25.1, as RNAi studies have indicated roles for cdc-25.1 during female meiosis and embryonic divisions (Ashcroft et al., 1999; Clucas et al., 2002).
For both cdc-25.1 and wee-1.3, gain-of-function mutants have been identified with surprising lineage-specific defects. Two independent dominant mutations in cdc-25.1 induce extra divisions in the intestine (Clucas et al., 2002; Kostic and Roy, 2002). Gain-of-function alleles of spe-37/wee-1.3 Myt1 cause spermatogenesis defects, resulting from G2/M arrest during male meiosis (Lamitina and L'Hernault, 2002). The molecular consequences of these mutations are not precisely known, but the phenotypes indicate either a different CDK-activity threshold for specific developmental decisions, or differential regulation of CDKs within certain lineages.
Which CDKs are regulated by inhibitory phosphorylation in C. elegans? The ATP-binding sites of CDK-1 and K03E5.3 each contain adjoining threonine and tyrosine residues, while CDK-4 contains alanine and tyrosine at the corresponding positions, similar to vertebrate Cdk4/Cdk6. It is likely that the dominant wee-1.3 mutations affect CDK-1, while cdc-25.1 gain of function may cause premature activation of a G1 CDK/Cyclin complex.
In addition to inhibition, CDKs are also positively regulated by phosphorylation. Activation of the CDK/Cyclin complex requires phosphorylation of a specific threonine residue in the activation loop. Based on results in other species, CDK-7 was examined as the candidate positive regulator (Wallenfang and Seydoux, 2002). Indeed, combined inactivation of cdk-7 through a temperature-sensitive mutation and RNAi resulted in a one-cell arrest, similar to cdk-1(RNAi) embryos. These results indicate strongly that CDK-7 kinase activity is required for CDK-1 activation (see also: CDKs and cyclins).
Protein degradation through ubiquitin-mediated proteolysis plays an important role in cell-cycle regulation. In C. elegans, this was first established through the characterization of lin-19/cul-1 and lin-23, which were originally defined in screens for mutants with abnormal postembryonic cell lineages. Blast cells undergo excessive divisions in all post-embryonic lineages in lin-19/cul-1 and lin-23 loss-of-function mutants (Kipreos et al., 2000; Kipreos et al., 1996). Cells divide at the appropriate times in these mutants, but additional cycles continue when cells should terminally differentiate. Thus, cul-1 and lin-23 act to promote cell-cycle exit.
lin-19 is the founding member of the conserved cullin gene family and was renamed "cul-1" (Kipreos et al., 1996). lin-23 encodes an F-box/WD-repeat protein, most similar to MET30 in yeast, human β-TRCP and Slmb in Drosophila (Kipreos et al., 2000). Based on the similar loss-of-function phenotype and molecular identity, CUL-1 and LIN-23 probably encode components of a Skp1-Cul1-F box (SCF) protein complex that acts as an E3 ubiquitin ligase and targets proteins for degradation (see Ubiquitin-mediated pathways in C. elegans ). This complex likely promotes cell-cycle arrest during larval development by promoting destruction of positive G1/S regulators. CYE-1 Cyclin E is a candidate critical target of CUL-1/LIN-23 SCF, analogous to the role of vertebrate Cul1 SCF.
The cullin family is comprised of six different genes in C. elegans and cell-cycle related functions have also been reported for three additional members. Deletion of cul-2 results in G1 arrest of germ-precursor cells, while cul-2(RNAi) embryos show defects in chromosome condensation and segregation (Feng et al., 1999) and are defective in progression through meiotic anaphase II (Liu et al., 2004; Sonneville and Gonczy, 2004). CUL-3 is specifically required for degradation of MEI-1, a subunit of a katanin complex that severs microtubules and is essential for meiotic chromosome segregation (Furukawa et al., 2003; Pintard et al., 2003; van den Heuvel, 2004; Xu et al., 2003). Finally, cul-4 is essential to prevent re-replication of DNA, likely by promoting degradation of the replication licencing factor CDT-1 (Zhong et al., 2003). These results demonstrate the importance of various cullin-based ubiquitin ligases in cell-cycle progression.
The anaphase-promoting complex (APC) is another multi-subunit E3 ubiquitin ligase that regulates cell-cycle transitions. As in other eukaryotes, C. elegans APC promotes sister chromosome separation in M phase. emb-30 is required for the metaphase-to-anaphase transition in meiosis and mitosis and encodes the APC subunit APC4 (Furuta et al., 2000). Moreover, a screen for temperature-sensitive embryonic lethal mutations revealed a large number of metaphase-to-anaphase transition (mat) mutants (Golden et al., 2000). These mat mutants arrest in metaphase of meiosis I and helped define five different genes that all encode components of the APC (Table 1).
In other species, the adaptor Cdc20/Fizzy recruits the APC to the substrate securin. Degradation of securin releases separase, which cleaves cohesins and triggers sister chromosome separation. Similar mechanisms are likely used in C. elegans, involving FZY-1 as the substrate specificity factor homologous to Cdc20/Fizzy, the interactor of FZY-1, as candidate securin and SEP-1 as the C. elegans separase (Kitagawa et al., 2002). The APC uses another substrate specificity factor, known as Cdh1p in yeast and Fizzy-related in Drosophila, in the destruction of mitotic cyclins. Loss of function of fzr-1, the C. elegans Cdh1/FZR ortholog, promotes hyperproliferation when combined with lin-35 Rb loss of function (Fay et al., 2002). This observation may indicate that FZR-1 also contributes to G1 cyclin degradation, or, alternatively, it may counteract CKI-1 Kip1 degradation. The latter mechanism has been observed in human cells, in which Cdh1/FZR promotes accumulation of p27Kip1 through degradation of the F box factor that targets p27Kip1 for destruction (Bashir et al., 2004; Wei et al., 2004).
As metazoans go through development, their cells progress through various types of cell cycles. These include the embryonic cell cycle, somatic cell cycle, endoreduplication cycle, and meiotic cell cycle. The cell-cycle machinery used in each case is tailored towards the individual cycle and shows different requirements for critical regulators. For instance, C. elegans cyd-1 cyclin D and cdk-4 Cdk4/6 are not required for most of embryogenesis and cdk-1 is not needed for the endoreduplication cycles (Boxem et al., 1999; Boxem and van den Heuvel, 2001; Park and Krause, 1999). Switching from one division cycle to another involves important developmental decisions that remain poorly understood.
As in other metazoans, early embryonic divisions in C. elegans are fast and cycle between S and M phases, apparently lacking the Gap phases G1 and G2 (Edgar and McGhee, 1988). In the initial division cycles, DNA synthesis, nuclear division, and cytokinesis are completed within approximately 15-20 minutes. However, the exact division times vary, as even the first mitotic division is asymmetric and generates daughter cells that are unequal and divide asynchronously. Just a few hours into embryonic development, cells in different lineages diverge greatly in cell-cycle profiles. Certain cells continue rapid divisions, others divide after an extended interphase of two hours or more, yet other cells become quiescent or post-mitotic (Sulston et al., 1983). Nearly all embryonic divisions are completed during the first half of embryogenesis, within the proliferation phase that ends approximately 7 hours after fertilization.
Because of the variation in cell division profiles, C. elegans embryogenesis does not include a clear switch from cycles that consist solely of S and M phases, to cycles that incorporate a G2 phase, or a G1 and G2 phase. The time of introduction of Gap phases depends on the lineage, with the endoderm precursors Ea and Ep as the first cells to include a G2 phase in their cycles at the 28-cell stage (Edgar and McGhee, 1988). These intestinal cells complete S phase before they start inward migration during gastrulation and divide approximately 1 hour later. When G1 is first introduced is unclear. As the final embryonic divisions of the intestinal and coelomocyte precursors fail in cyd-1 mutants (Boxem and van den Heuvel, 2001; Yanowitz and Fire, 2005), these cycles most likely include G1 phases.
The somatic nuclei of post-embryonic precursor cells appear to contain a 2n DNA content at the time of hatching and go through a DNA synthesis phase before initiating mitosis (Albertson et al., 1978; Hedgecock and White, 1985). These divisions generally depend on the function of G1/S and G2/M control genes. Measurements of the time of DNA duplication in the vulval precursor cells demonstrated that G1/G0 extends from mid L1 until shortly after the L2 molt, and that S phase is completed hours before mitosis initiates (Euling and Ambros, 1996b). Similarly, S phase in the intestinal nuclei occurs between 6 and 8 hours of L1 development, approximately 4 hours before nuclear division (Boxem and van den Heuvel, 2001). Thus, the precursor cells of the post-embryonic lineages and their descendents follow canonical cell cycles in which the S and M phases are separated by G1 and G2 Gap phases. As in embryogenesis, the length of interphase varies greatly between different cell types. Divisions frequently follow each other within one hour, but some cells remain quiescent for 20 hours, before dividing again two larval stages later (Sulston and Horvitz, 1977).
Endoreduplication cycles are characterized by a DNA synthesis phase that is not followed by M phase, thus doubling the DNA ploidy with each additional cycle. Such endoreduplication cycles take place in the intestine and hypodermis during C. elegans development (Hedgecock and White, 1985). Fourteen of the twenty intestinal cells undergo a final nuclear division at the end of the L1 stage. Subsequently, all intestinal nuclei go through an endoreduplication cycle during each larval stage, which results in intestinal nuclei with a 32n DNA content in adult animals.
During the larval stages, divisions of the hypodermal seam cells generate daughter seam cells as well as hypodermal cells that fuse with the major hypodermal syncytium hyp7 (Sulston and Horvitz, 1977). The cells that become part of hyp7 undergo an additional round of DNA replication just before they join the syncytium (Hedgecock and White, 1985). Consequently, the larval hyp7 syncytium contains a fixed number of diploid embryonic nuclei and an increasing amount of tetraploid postembryonic nuclei.
In meiosis, DNA synthesis is followed by two subsequent rounds of chromosome segregation, leading to the formation of haploid gametes (see Introduction to the germ line). Hermaphrodites temporarily produce male gametes during the third larval stage, before switching to an oogenesis program. In adult animals, a proliferating stem-cell population at the distal end of each gonad arm forms precursor germ cells. These precursor cells go through S phase and enter a prolonged meiotic prophase, in which homologous chromosomes pair, synapse and undergo recombination in the pachytene stage. Oocytes complete development while in diakinesis, and undergo maturation when reaching the spermatheca. The oocyte pronucleus completes meiosis I and II upon fertilization. Meiosis is described elsewhere (see Meiosis).
Checkpoint controls prevent progression through cell-cycle transitions prior to the completion of critical earlier events. For instance, the cell cycle is arrested and cell death may be triggered in response to DNA damage, progress into mitosis is halted when DNA replication is ongoing, and sister-chromatid separation is delayed until all kinetochores are attached to the spindle. The DNA damage checkpoint, replication checkpoint and spindle-assembly checkpoint, respectively, impose these "brakes" on the cell cycle and couple the initiation of later events to completion of earlier events. Recent studies in C. elegans have demonstrated conservation of these checkpoint pathways, identified novel components and revealed developmental functions.
While somatic cells show little developmental response to genotoxic stress, DNA damage in the germline induces both cell-cycle arrest and apoptosis (Gartner et al., 2000). Cell-cycle arrest is restricted to the mitotically active stem-cell population in the distal region of the gonad arms, while cell death occurs only in the meiotic pachytene regions in adult hermaphrodites. As in other eukaryotes, the mrt-2 RAD1 and hus-1 HUS1 genes are required for checkpoint-induced arrest as well as apoptosis, together with the novel checkpoint gene clk-2/rad-5 (Ahmed et al., 2001). It is currently unclear how damage triggers cell-cycle arrest in C. elegans. Neither cki-1 nor cki-2 Cip/Kip expression is induced in response to irradiation, which rules out one of the mechanisms used in mammals (Hofmann et al., 2002). Other conserved mechanisms involve inactivation of Cdc25 and activation of Wee1 family members, which may mediate the arrest in C. elegans.
As DNA damage induces apoptosis in metazoans but not in yeast, C. elegans provides an attractive genetic system to establish the molecular connections. Importantly, a p53-related transcription factor (CEP-1: C. elegans p53) acts to promote checkpoint-induced apoptosis in C. elegans, as in mammals (Derry et al., 2001; Schumacher et al., 2001). In response to gamma irradiation, transcript levels of egl-1 and ced-13 are both induced in a cep-1 dependent fashion (Hofmann et al., 2002; Schumacher et al., 2005). EGL-1 and CED-13 each encode pro-apoptotic proteins of the BH3-only protein family, similar to the p53 targets Bax, Puma and Noxa in mammals. These results highlight the evolutionary conservation of the damage-induced apoptotic pathway (see Germline survival and cell death ).
As in other eukaryotes, interference of DNA replication by Hydroxyurea (HU) or mutation of replication components prevents progression into mitosis (Encalada et al., 2000; Euling and Ambros, 1996b). Interestingly, the replication checkpoint responsible for this delay is also active in early embryos, albeit less robustly (Brauchle et al., 2003). RNAi of the replication-checkpoint genes atl-1 ATR and chk-1 CHK1 by RNAi resulted in precocious and more synchronous division of the AB and P1 blastomeres. Thus, the replication checkpoint contributes to the normal and lineage specific timing of cell division in early development.
Spindle checkpoint components were originally identified in yeast as "mitotic arrest deficient" (mad) and "budding uninhibited by benzimidazole" (bub) mutants. The C. elegans gene mdf-2 (mitotic arrest defective) was identified as a homolog of MAD2 and shown to be able to substitute for its function (Kitagawa and Rose, 1999). MDF-1 was identified as a protein interacting with MDF-2 that weakly resembles Mad1p and produces a similar range of loss-of-function phenotypes as mdf-2, including embryonic lethality, larval lethality, a Him phenotype and sterility. These results provided the first indication that mitotic checkpoint genes are essential in metazoans.
The existence of a functional spindle checkpoint was demonstrated by exposure of the germline to the microtubule-depolymerizing drug nocodazole, which causes accumulation of cells in mitosis (Kitagawa and Rose, 1999). Such accumulation does not occur in mdf-1 mutants, indicating that mdf-1 is required for mitotic arrest in response to spindle defects. This mitotic checkpoint is also functional in early embryos, as defective mitotic spindles expand the time in mitosis in a checkpoint dependent manner as early as the one- and two-cell stage (Encalada et al., 2005).
Other mitotic checkpoint proteins are also conserved in C. elegans, including orthologs of BUB1 and MAD3. Interestingly, san-1 MAD3 was identified as a gene required for an extreme form of developmental quiescence induced by absence of oxygen (anoxia) (Nystul et al., 2003). This so-called "suspended animation" coincides with mitotic metaphase arrest during embryogenesis. Not only SAN-1 MAD3 but also MDF-2 MAD2 was found to be required for this arrest induced by adverse conditions.
At present, there is only limited understanding of developmental regulation of cell cycle entry and arrest in C. elegans. Several environmental conditions and mutations cause a global interruption of the reproducible pattern of cell division. For instance, when eggs hatch in the absence of food, larvae arrest early in L1 and do not initiate development and cell division until food is restored. Similarly, entry into and exit from the dauer state coincide with arrest and re-initiation of cell division. In addition, heterochronic mutations affect the timing of cell division simultaneously in multiple postembryonic lineages.
It is unclear how such developmental programs control the cell cycle and whether systemic signals are involved. Most likely however, regulation occurs via the cyclin/Cdk complexes. Inactivation of cyd-1 and cdk-4 leads to G1 arrest (see above, CDKs and cyclins; Figure 3), and heat-shock induced expression of cyd-1 plus cdk-4 is sufficient to induce expression of the S phase marker rnr::GFP in L1 animals arrested through starvation (Park and Krause, 1999). Conversely, ectopic expression of the CDK inhibitor CKI-1 induces cell-cycle arrest and prevents rnr::gfp expression, while cki-1 loss-of-function results in extra cell division (see: CDK inhibitory proteins). RNAi of cki-1 induces rnr::gfp expression and limited cell division even in starved L1 animals and arrested dauer larvae (Hong et al., 1998). Mutations in heterochronic genes that cause precocious or retarded patterns of cell division affect the time cells spend in G0/G1, probably also acting upstream of the basic cell cycle machinery (Euling and Ambros, 1996b). Thus, both the normal developmental timing of cell division and environmental, possibly systemic, regulation is likely accomplished through regulators acting upstream of G1 cyclin/CDK complexes.
What then controls the proper timing of cell-cycle entry and arrest? Promoter studies have shown that cyd-1, cdk-4 and cye-1 all are transcriptionally induced in dividing cells (Brodigan et al., 2003; Park and Krause, 1999). However, homozygous cye-1 mutants show normal temporal control of cell division during embryonic and early larval development, illustrating that zygotic cye-1 transcription does not drive lineage-specific timing of cell-cycle entry (Brodigan et al., 2003; Fay and Han, 2000). In contrast to cyd-1 and cye-1, the cki-1 promoter region is large and complex, and includes separate control elements for transcription in different lineages (Hong et al., 1998). Thus, transcription of cki-1 likely contributes to timing of cell cycle arrest.
Another level of CKI-1 regulation may be mediated by phosphorylation. Just like CKI-1, the CDC-14 phosphatase is needed to maintain the vulval precursor cells (VPCs) and other postembryonic precursor cells in a resting state (Saito et al., 2004). In genetic studies, cdc-14 was found to act positively in a cki-1 pathway. Moreover, a CKI-1::GFP fusion protein, expressed from a heterologous promoter, failed to accumulate in the absence of cdc-14 function. Based on analogy with other systems, CKI-1 turn-over is likely promoted by phosphorylation and ubiquitin-dependent proteolysis. CDC-14 phosphatase activity could contribute to cell-cycle arrest by counteracting CKI-1 phosphorylation, thereby preventing CKI-1 degradation.
Most cells exit the cell cycle and differentiate appropriately in cki-1 mutants, and even in animals that lack both lin-35 and cki-1 function. Cell-cycle exit is delayed in lin-23 and cul-1 mutants, but the smaller extra cells still differentiate (Kipreos et al., 1996; Kipreos and Pagano, 2000). Thus, cells can transit from G1 to G0 even when these negative regulators of G1 progression are missing, possibly because redundant mechanisms are in place.
Differentiation is often surprisingly independent from cell division. Even in the absence of larval divisions, cyd-1 mutants and cdk-1 mutant animals continue molting cycles and form adult alae. These observations confirm earlier conclusions that aspects of development and differentiation can continue in the absence of cell division (Albertson et al., 1978). Vulval cells in cye-1 mutants undergo normal differentiation and morphogenesis of the vulva, despite delayed and fewer division cycles (Fay and Han, 2000). Extra divisions of the VPCs, in cki-1(RNAi) and cdc-14(0) larvae, or extra vulval cells, in cul-1 and lin-23 mutants, also does not prevent acquisition of a normal vulval fate or vulval morphogenesis (Hong et al., 1998; Kipreos et al., 2000; Kipreos et al., 1996; Saito et al., 2004).
Despite this independence, receptiveness and response to cell-cell signaling does include a cell-cycle component. The number of VPCs adopting a vulval fate is somewhat increased in cye-1 mutants, possibly because prolonged time in G1 provides greater opportunity for induction (Fay and Han, 2000). Different VPC fates may use a different "window of opportunity" for lin-12-mediated lateral inhibition, with the 1° versus 2° decision requiring signaling earlier than the 2° versus 3° decision (Ambros, 1999). In precocious lin-28 mutants, VPCs adopt vulval fates and initiate fate-specific division patterns in L2. Dauer induction interrupts this pattern and arrests the VPC daughter cells (Euling and Ambros, 1996a). Interestingly, these arrested cells lose their lineage commitment and respond as multipotent VPCs to vulval induction during post-dauer development. In all three examples, the time spent in G1 affects the VPC fate in response to signals.
Developmental decisions and cell-cycle regulation interconnect at many levels and much remains to be discovered. Germline proliferation and its control describes proliferation in the germline, which involves some of the best characterized connections between developmental signals and cell-division control.
I thank Mike Boxem, John Koreth and Jerome Korzelius for helpful comments and discussion, Edward Kipreos for critical reading and helpful suggestions, and the National Institutes of Health for support.
Ahmed, S., Alpi, A., Hengartner, M.O., and Gartner, A. (2001). C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr. Biol. 11, 1934–1944. Abstract Article
Albertson, D.G., Sulston, J.E., and White, J.G. (1978). Cell cycling and DNA replication in a mutant blocked in cell division in the nematode Caenorhabditis elegans. Dev. Biol. 63, 165–178. Abstract Article
Ambros, V. (1999). Cell cycle-dependent sequencing of cell fate decisions in Caenorhabditis elegans vulva precursor cells. Development 126, 1947–1956. Abstract
Ashcroft, N., and Golden, A. (2002). CDC-25.1 regulates germline proliferation in Caenorhabditis elegans. Genesis 33, 1–7. Abstract Article
Ashcroft, N.R., Srayko, M., Kosinski, M.E., Mains, P.E., and Golden, A. (1999). RNA-mediated interference of a cdc25 homolog in Caenorhabditis elegans results in defects in the embryonic cortical membrane, meiosis, and mitosis. Dev. Biol. 206, 15–32. Abstract Article
Bashir, T., Dorrello, N.V., Amador, V., Guardavaccaro, D., and Pagano, M. (2004). Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428, 190–193. Abstract Article
Boxem, M., Srinivasan, D.G., and van den Heuvel, S. (1999). The Caenorhabditis elegans gene ncc-1 encodes a cdc2-related kinase required for M phase in meiotic and mitotic cell divisions, but not for S phase. Development 126, 2227–2239. Abstract
Boxem, M., and van den Heuvel, S. (2001). lin-35 Rb and cki-1 Cip/Kip cooperate in developmental regulation of G1 progression in C. elegans. Development 128, 4349–4359. Abstract
Boxem, M., and van den Heuvel, S. (2002). C. elegans class B synthetic multivulva genes act in G(1) regulation. Curr. Biol. 12, 906–911. Abstract Article
Brauchle, M., Baumer, K., and Gonczy, P. (2003). Differential activation of the DNA replication checkpoint contributes to asynchrony of cell division in C. elegans embryos. Curr. Biol. 13, 819–827. Abstract Article
Brodigan, T.M., Liu, J., Park, M., Kipreos, E.T., and Krause, M. (2003). Cyclin E expression during development in Caenorhabditis elegans. Dev. Biol. 254, 102–115. Abstract Article
Clucas, C., Cabello, J., Bussing, I., Schnabel, R., and Johnstone, I.L. (2002). Oncogenic potential of a C. elegans cdc25 gene is demonstrated by a gain-of-function allele. EMBO J. 21, 665–674. Abstract Article
Cruz, J.C., and Tsai, L.H. (2004). Cdk5 deregulation in the pathogenesis of Alzheimer's disease. Trends Mol. Med. 10, 452–458. Abstract Article
Datar, S.A., Jacobs, H.W., de la Cruz, A.F., Lehner, C.F., and Edgar, B.A. (2000). The Drosophila cyclin D-Cdk4 complex promotes cellular growth. EMBO J. 19, 4543–4554. Abstract Article
Derry, W.B., Putzke, A.P., and Rothman, J.H. (2001). Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294, 591–595. Abstract Article
Edgar, L.G., and McGhee, J.D. (1988). DNA synthesis and the control of embryonic gene expression in C. elegans. Cell 53, 589–599. Abstract Article
Encalada, S.E., Martin, P.R., Phillips, J.B., Lyczak, R., Hamill, D.R., Swan, K.A., and Bowerman, B. (2000). DNA replication defects delay cell division and disrupt cell polarity in early Caenorhabditis elegans embryos. Dev. Biol. 228, 225–238. Abstract Article
Encalada, S.E., Willis, J., Lyczak, R., and Bowerman, B. (2005). A spindle checkpoint functions during mitosis in the early Caenorhabditis elegans embryo. Mol. Biol. Cell 16, 1056–1070. Abstract Article
Euling, S., and Ambros, V. (1996a). Heterochronic genes control cell cycle progress and developmental competence of C. elegans vulva precursor cells. Cell 84, 667–676. Abstract Article
Euling, S., and Ambros, V. (1996b). Reversal of cell fate determination in Caenorhabditis elegans vulval development. Development 122, 2507–2515. Abstract
Fay, D.S., and Han, M. (2000). Mutations in cye-1, a Caenorhabditis elegans cyclin E homolog, reveal coordination between cell-cycle control and vulval development. Development 127, 4049–4060. Abstract
Fay, D.S., Keenan, S., and Han, M. (2002). fzr-1 and lin-35/Rb function redundantly to control cell proliferation in C. elegans as revealed by a nonbiased synthetic screen. Genes Dev. 16, 503–517. Abstract Article
Feng, H., Zhong, W., Punkosdy, G., Gu, S., Zhou, L., Seabolt, E.K., and Kipreos, E.T. (1999). CUL-2 is required for the G1-to-S-phase transition and mitotic chromosome condensation in Caenorhabditis elegans. Nat. Cell Biol. 1, 486–492. Abstract Article
Fisher, R.P., and Morgan, D.O. (1994). A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell 78, 713–724. Abstract Article
Fukuyama, M., Gendreau, S.B., Derry, W.B., and Rothman, J.H. (2003). Essential embryonic roles of the CKI-1 cyclin-dependent kinase inhibitor in cell-cycle exit and morphogenesis in C. elegans. Dev. Biol. 260, 273–286. Abstract Article
Furukawa, M., He, Y.J., Borchers, C., and Xiong, Y. (2003). Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat. Cell Biol. 5, 1001–1007. Abstract Article
Furuta, T., Tuck, S., Kirchner, J., Koch, B., Auty, R., Kitagawa, R., Rose, A.M., and Greenstein, D. (2000). EMB-30: an APC4 homologue required for metaphase-to-anaphase transitions during meiosis and mitosis in Caenorhabditis elegans. Mol. Biol. Cell 11, 1401–1419. Abstract
Gartner, A., Milstein, S., Ahmed, S., Hodgkin, J., and Hengartner, M.O. (2000). A conserved checkpoint pathway mediates DNA damage--induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443. Abstract Article
Golden, A., Sadler, P.L., Wallenfang, M.R., Schumacher, J.M., Hamill, D.R., Bates, G., Bowerman, B., Seydoux, G., and Shakes, D.C. (2000). Metaphase to anaphase (mat) transition-defective mutants in Caenorhabditis elegans. J. Cell Biol. 151, 1469–1482. Abstract Article
Hedgecock, E.M., and White, J.G. (1985). Polyploid tissues in the nematode Caenorhabditis elegans. Dev. Biol. 107, 128–133. Abstract Article
Hofmann, E.R., Milstein, S., Boulton, S.J., Ye, M., Hofmann, J.J., Stergiou, L., Gartner, A., Vidal, M., and Hengartner, M.O. (2002). Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 12, 1908–1918. Abstract Article
Hong, Y., Roy, R., and Ambros, V. (1998). Developmental regulation of a cyclin-dependent kinase inhibitor controls postembryonic cell cycle progression in Caenorhabditis elegans. Development 125, 3585–3597. Abstract
Kipreos, E.T., Gohel, S.P., and Hedgecock, E.M. (2000). The C. elegans F-box/WD-repeat protein LIN-23 functions to limit cell division during development. Development 127, 5071–5082. Abstract
Kipreos, E.T., Lander, L.E., Wing, J.P., He, W.W., and Hedgecock, E.M. (1996). cul-1 is required for cell cycle exit in C. elegans and identifies a novel gene family. Cell 85, 829–839. Abstract Article
Kipreos, E.T., and Pagano, M. (2000). The F-box protein family. Genome Biol. 1, REVIEWS3002. Abstract
Kitagawa, R., Law, E., Tang, L., and Rose, A.M. (2002). The Cdc20 homolog, FZY-1, and its interacting protein, IFY-1, are required for proper chromosome segregation in Caenorhabditis elegans. Curr. Biol. 12, 2118–2123. Abstract Article
Kitagawa, R., and Rose, A.M. (1999). Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat. Cell Biol. 1, 514–521. Abstract Article
Kostic, I., Li, S., and Roy, R. (2003). cki-1 links cell division and cell fate acquisition in the C. elegans somatic gonad. Dev. Biol. 263, 242–252. Abstract Article
Kostic, I., and Roy, R. (2002). Organ-specific cell division abnormalities caused by mutation in a general cell cycle regulator in C. elegans. Development 129, 2155–2165. Abstract
Kreutzer, M.A., Richards, J.P., De Silva-Udawatta, M.N., Temenak, J.J., Knoblich, J.A., Lehner, C.F., and Bennett, K.L. (1995). Caenorhabditis elegans cyclin A- and B-type genes: a cyclin A multigene family, an ancestral cyclin B3 and differential germline expression. J. Cell Sci. 108 (Pt 6), 2415-2424. Abstract
Lamitina, S.T., and L'Hernault, S.W. (2002). Dominant mutations in the Caenorhabditis elegans Myt1 ortholog wee-1.3 reveal a novel domain that controls M-phase entry during spermatogenesis. Development 129, 5009–5018. Abstract
Liu, J., and Kipreos, E.T. (2000). Evolution of cyclin-dependent kinases (CDKs) and CDK-activating kinases (CAKs): differential conservation of CAKs in yeast and metazoa. Mol. Biol. Evol. 17, 1061–1074. Abstract
Liu, J., Vasudevan, S., and Kipreos, E.T. (2004). CUL-2 and ZYG-11 promote meiotic anaphase II and the proper placement of the anterior-posterior axis in C. elegans. Development 131, 3513–3525. Abstract Article
Meyer, C.A., Jacobs, H.W., Datar, S.A., Du, W., Edgar, B.A., and Lehner, C.F. (2000). Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 19, 4533–4542. Abstract Article
Myers, T.R., and Greenwald, I. (2005). lin-35 Rb acts in the major hypodermis to oppose ras-mediated vulval induction in C. elegans. Dev. Cell 8, 117–123. Abstract Article
Mori, H., Palmer, R.E., and Sternberg, P.W. (1994). The identification of a Caenorhabditis elegans homolog of p34cdc2 kinase. Mol. Gen Genet. 245, 781–786. Abstract Article
Nystul, T.G., Goldmark, J.P., Padilla, P.A., and Roth, M.B. (2003). Suspended animation in C. elegans requires the spindle checkpoint. Science 302, 1038–1041. Abstract Article
Park, M., and Krause, M.W. (1999). Regulation of postembryonic G(1) cell cycle progression inCaenorhabditis elegans by a cyclin D/CDK-like complex. Development 126, 4849–4860. Abstract
Pintard, L., Willis, J.H., Willems, A., Johnson, J.L., Srayko, M., Kurz, T., Glaser, S., Mains, P.E., Tyers, M., Bowerman, B. et al. (2003). The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature 425, 311–316. Abstract Article
Saito, R.M., Perreault, A., Peach, B., Satterlee, J.S., and van den Heuvel, S. (2004). The CDC-14 phosphatase controls developmental cell-cycle arrest in C. elegans. Nat. Cell Biol. 6, 777–783. Abstract Article
Schumacher, B., Hofmann, K., Boulton, S., and Gartner, A. (2001). The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 11, 1722–1727. Abstract Article
Schumacher, B., Schertel, C., Wittenburg, N., Tuck, S., Mitani, S., Gartner, A., Conradt, B., and Shaham, S. (2005). C. elegans ced-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ 12, 153–161. Abstract Article
Sherr, C.J., and Roberts, J.M. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512. Abstract
Shiekhattar, R., Mermelstein, F., Fisher, R.P., Drapkin, R., Dynlacht, B., Wessling, H.C., Morgan, D.O., and Reinberg, D. (1995). Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature 374, 283–287. Abstract Article
Shim, E.Y., Walker, A.K., Shi, Y., and Blackwell, T.K. (2002). CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev. 16, 2135–2146. Abstract Article
Sonneville, R., and Gonczy, P. (2004). zyg-11 and cul-2 regulate progression through meiosis II and polarity establishment in C. elegans. Development 131, 3527–3543. Abstract Article
Stevaux, O., and Dyson, N.J. (2002). A revised picture of the E2F transcriptional network and RB function. Curr. Opin. Cell Biol. 14, 684–691. Abstract Article
Sulston, J.E., and Horvitz, H.R. (1977). Post-embryonic cell lineages of the nematode,Caenorhabditis elegans. Dev. Biol. 56, 110–156. Abstract Article
Sulston, J.E., Schierenberg, E., White, J.G., and Thomson, J.N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64–119. Abstract Article
van den Heuvel, S. (2004). Protein degradation: CUL-3 and BTB--partners in proteolysis. Curr. Biol. 14, R59–R61. Abstract Article
Wallenfang, M.R., and Seydoux, G. (2002). cdk-7 is required for mRNA transcription and cell cycle progression in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. USA 99, 5527–5532. Abstract Article
Wei, W., Ayad, N.G., Wan, Y., Zhang, G.J., Kirschner, M.W., and Kaelin, W.G., Jr. (2004). Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 428, 194–198. Abstract Article
Xu, L., Wei, Y., Reboul, J., Vaglio, P., Shin, T.H., Vidal, M., Elledge, S.J., and Harper, J.W. (2003). BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature 425, 316–321. Abstract Article
*Edited by James M. Kramer and Donald G. Moerman. Last revised May 13, 2005. Published September 21, 2005. This chapter should be cited as: van den Heuvel, S. Cell-cycle regulation (September 21, 2005), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.28.1, http://www.wormbook.org.
Copyright: © 2005 Sander van den Heuvel. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: heuvel@helix.mgh.harvard.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.