Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Autophagy is a ubiquitous cellular process responsible for the bulk degradation of cytoplasmic components through an autophagosomal-lysosomal pathway. Genetic screens, primarily in S. cerevisiae, have identified numerous genes that are essential for autophagy. Many of these genes have orthologs in higher eukaryotes, including C. elegans, Drosophila, and mammals. Gene knockdown/knockout studies in C. elegans have been useful to probe the functions of autophagy in an intact multicellular organism that undergoes development to produce different cell types. This review summarizes important themes that have emerged regarding the roles of autophagy in C. elegans in adaptation to stress, aging, normal reproductive growth, cell death, cell growth control, neural synaptic clustering, and the degradation of aggregate-prone proteins.
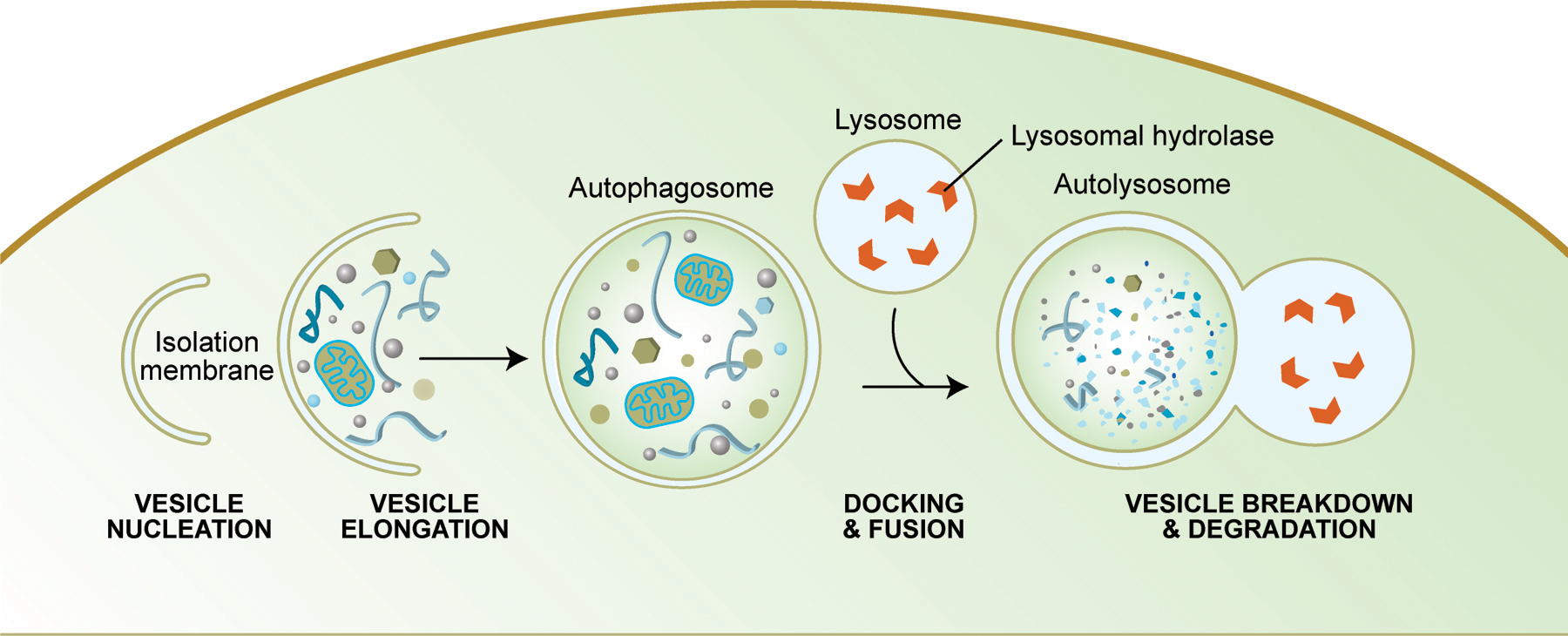
The term autophagy literally means “self-eating” and denotes any cellular pathway involving the delivery of cytoplasmic material to the lysosome for degradation. There are at least three types of autophagy, chaperone-mediated autophagy, microautophagy and macroautophagy, which differ with respect to the mode of delivery to the lysosome. Chaperone-mediated autophagy is a mechanism that allows the direct lysosomal import of proteins which contain a particular pentapeptide motif (Majeski and Dice, 2004; Massey et al., 2004), whereas both microautophagy and macroautophagy involve dynamic membrane rearrangements that terminate at the lysosome. In microautophagy, cytoplasmic material is directly engulfed at the surface of the lysosome by septation, protrusion or invagination of the limiting membrane. Macroautophagy involves the sequestration of cytoplasm into a double-membrane cytosolic vesicle referred to as an autophagosome, that subsequently fuses with a late endosome or lysosome to form an autophagolysosome (or autolysome). Inside the autophagolysosome, the lysosomal hydrolases degrade the sequestered material, which then becomes available to the cell for recycling. Macroautophagy is the major regulated cellular pathway for degrading long-lived proteins and the only known pathway for degrading cytoplasmic organelles (reviewed in Dunn, 1994; Klionsky, 2007; Klionsky and Emr, 2000).
How autophagosomes are formed remains somewhat controversial because the origin of the membrane is unknown. In Saccharomyces cerevisiae, the initial step of autophagy consists of the formation of a phagopore, or isolation membrane (vesicle nucleation step). The proposed site for autophagosome formation is the phagopore assembly site (PAS) (Kim et al., 2002; Kirisako et al., 1999; Suzuki et al., 2001). This structure consists of the phagopore, or its precursor, and the autophagy proteins that form the core machinery of the forming autophagosome. The concerted action of the autophagy core machinery proteins at the PAS is thought to lead to the expansion of the phagopore into an autophagosome. In mammalian systems, it is not yet known whether the PAS exists, although colocalization of autophagy core machinery proteins to the phagopore has also been observed (Mizushima et al., 2003; Mizushima et al., 2001). Fusion of the phagopore edges forms the autophagosome during the vesicle elongation and completion steps. Docking and fusion occurs when the outer membrane of the autophagosome fuses with the vacole (in yeast) or the lysosome (in mammalian cells), producing an autophagolysosome in mammals, and an autophagic body in yeast. Finally, the sequestered material is degraded by vacuolar or lysosomal hydrolases inside the autophagosome, and the degradation products are reutilized by the cell.
In this review we focus on macroautophagy (herein referred to as autophagy) as virtually all of the studies in C. elegans have centered on this form of autophagy. Investigations of the cellular events involved in autophagy in C. elegans have been limited, but it is presumed that the formation and fate of the autophagosome is essentially similar to that in yeast and mammalian cells (Figure 1). There may, however, be some subtle differences between C. elegans and other species. For example, S. cerevisiae autophagosomes are usually about 400-900 nm in diameter (Takeshige et al., 1992), and mammalian autophagosomes are usually between 500-2000 nm, whereas C. elegans autophagosomes appear to be smaller, about 300-500 nm in diameter (Kovács, 2004; Meléndez et al., 2003). The reason for the size difference of autophagosomes is unknown. Given the ease of studying the cellular events of autophagy in yeasts or cultured mammalian cells (and the challenges that occur in addressing this area in the intact nematode), it is likely that future studies in C. elegans will continue to focus on the genetics of the autophagy pathway and its biological roles in metazoan development and biology.
|
Figure 1: Schematic diagram of the steps of autophagy. Autophagy begins with the formation of the phagophore or isolation membrane (vesicle nucleation step). The concerted action of the autophagy core machinery proteins at the phagophore assembly site (PAS) is thought to lead to the expansion of the phagophore into an autophagosome (vesicle elongation). The autophagosome can engulf bulk cytoplasm nonspecifically, including entire organelles, or target cargos specifically. When the outer membrane of the autophagosome fuses with an endosome (forming an amphisome before fusing with the lysosome) or directly with a lysosome (docking and fusion steps), it forms an autophagolysosome. Finally, the sequestered material is degraded inside the autophagolyosome (vesicle breakdown and degradation) and recycled.
As detailed in numerous reviews on yeast autophagy, several genetic screens led to the identification of autophagy-related genes, or ATG genes, in Saccharomyces cerevisiae, Hansenula polymorpha, and Pichia pastoris. The analyses of yeast atg mutants have provided the framework for dissecting the autophagic process into distinct steps, including (Fig. 1): (a) induction, (b) cargo selection and packaging, (c) vesicle nucleation, (d) vesicle expansion and completion, (e) retrieval, and (f) vesicle targeting, docking, and fusion (reviewed in Suzuki and Ohsumi, 2007; Xie and Klionsky, 2007). Gene products have been identified that act at each of these steps, and that are required for the formation of the sequestering membrane (Fig. 2).
Four enzymatic complexes are required for autophagosome formation (Fig. 2); a serine/threonine protein kinase complex that responds to upstream inhibitory signals like the TOR kinase and induces autophagic activity; a class III phosphatidylinositol 3-kinase (PI3K) lipid kinase enzymatic complex that acts in vesicle nucleation; two novel ubiquitin-like conjugation pathways that lead to vesicle expansion and completion; and a protein retrieval system. In yeast, the induction complex includes the kinase, Atg1, and its regulators, Atg13 and Atg17, and it may interact with Atg23 and the integral membrane protein, Atg9, to recruit membrane to the nascent phagophore (Fig. 2A). Vesicle nucleation requires a lipid kinase complex, including: Atg6, Atg14, Vps15 and the class III PI3K Vps34, which function to generate phosphatidylinositol 3-phosphate (PI3P) (Fig. 2B) (Kihara et al., 2001). This class III PI3K complex functions in the localization of other autophagy proteins to the forming autophagosome (Kihara et al., 2001). Two other enzymatic complexes are the Atg12 conjugation system (Atg12, Atg5, and Atg16), and the Atg8 lipidation system (Atg8, Atg3, and Atg7) which mediate vesicle expansion, and vesicle completion (Fig. 2C). The retrieval system includes Atg9 which is the only transmembrane protein in the core machinery of autophagy proteins. Atg9 functions in the shuttling of membrane from the PAS to non-PAS structures, a process required for the formation of the autophagosome (Reggiori and Klionsky, 2006; Reggiori et al., 2004; Yen and Klionsky, 2007). The efficient delivery of Atg9 to the PAS involves the transport factors Atg23 and Atg27 (Legakis et al., 2007; Tucker et al., 2003; Yen et al., 2007). Finally, the retrieval of Atg9 (Fig. 2D) from the PAS involves the Atg1 kinase complex, as well as Atg2 and Atg18, two interacting peripheral proteins (Reggiori et al., 2004; Shintani et al., 2001; Suzuki et al., 2007; Wang et al., 2001).
|
Figure 2: Schematic diagram of the presumed role for C. elegans autophagy proteins involved in the formation of an autophagosome.
A) Regulation of induction: In yeast, the Tor kinase and its effectors regulate the induction of autophagy. UNC-51 is the C. elegans Atg1 ortholog, however, it is not clear if there are similar regulatory proteins to Atg17 or Atg13.
B) Vesicle nucleation requires a lipid kinase complex, which includes the class III phosphatidylinositol 3-kinase (PI3K), Vps34 (in C. elegans LET-512). In yeast, Vps34 activation depends on its binding partners, Atg6, Atg14, and Vps15 (Kihara et al., 2001). In C. elegans, the ATG6 ortholog is bec-1, however, orthologs to ATG14 and VPS15 appear to be missing in the C. elegans genome. The interaction between the antiapoptotic protein CED-9 and BEC-1 is conserved in C. elegans (Takács-Vellai et al., 2005). A similar interaction between the mammalian proteins Bcl-2 and Beclin 1, inhibits autophagy (Maiuri et al., 2007a; Maiuri et al., 2007b; Pattingre et al., 2005).
C) Two novel ubiquitin-like conjugation pathways: the Atg12 conjugation system (Atg5, Atg12, and Atg16), and the Atg8 lipidation system (Atg8, Atg3, and Atg7) mediate vesicle expansion, and vesicle completion. Orthologs to all the members of these two complexes have been found in C. elegans, where they are named ATG-3, ATG-4, ATG-5, ATG-7, ATG-16, LGG-1/Atg8 and LGG-3/Atg12. In yeast, Atg8 undergoes two posttranslational processing events resulting in conjugation to phosphatidylethanolamine (PE) and recruitment to the PAS membrane. ATG-7 is an E1 ubiquitin activating enzyme required for the activation of of LGG-1. LGG-3-ATG-5 oligomerize with ATG-16 to allow for the formation of the multimeric complex. ATG-3 and ATG-10 are E2-like ubiquitin conjugating enzymes and ATG-4 is a cysteine protease. As for its yeast ortholog, LGG-1 appears to remain in the completed autophagosome and thus is an excellent marker for early and late autophagosomal structures.
D) The retrieval of the integral membrane protein ATG-9 from the phagophore assembly site (PAS) involves ATG-2 and ATG-18, two interacting peripheral proteins.
Fusion of the autophagosome vesicle requires proteins common to all pathways (not just macroautophagy) that terminate at the vacuole/lysosome. It has been proposed that autophagosomes fuse with endosomes and become amphisomes, before fusion with lysosomes, which may provide the nascent autophagosomes with machinery that can be used for fusion with lysosomes (Berg et al., 1998; Tooze et al., 1990). In mammals, autophagolysosomal fusion involves the integral membrane protein LAMP2 (Gonzalez-Polo et al., 2005; Nishino et al., 2000), and Rab7, a GTP-binding protein (Kimura et al., 2007). Fusion in yeast involves the Rab family GTPase Ypt7, the SNAREs, Vam3, Vam7, Vti1 and Ykt6, members of the HOPS complex, and two components, Ccz1 and Mon1, which act at the docking stage (Luzio et al., 2007). In yeast, the breakdown of the vesicle and degradation requires Atg15, a putative lipase (Teter et al., 2001). In mammals, degradation requires a series of lysosomal proteases (cathepsins B, D and L) (Shacka and Roth, 2005). Subsequent to degradation, the resulting monomeric units (e.g. amino acids) that have been generated can be exported to the cytosol for reuse.
Many of the yeast genes encoding autophagy proteins have orthologs in the C. elegans genome (Table 1), for example: ATG2, ATG3, ATG4, ATG5, ATG6, ATG7, ATG8, ATG9, ATG10, ATG12, ATG16, ATG18, have been found in the C. elegans genome. Autophagy function has been documented for genes acting in autophagy induction (unc-51/ATG1), vesicle nucleation (bec-1/ATG6, vps-34/VPS34), the protein conjugation systems (atg-7/M7.5/ATG7, lgg-1/ATG8, lgg-3/ATG12), retrieval and vesicle recycling (atg-18/F41E6.13/ATG18) (Hansen et al., 2008; Hars et al., 2007; Meléndez et al., 2003). When searching for structural homology between yeast and C. elegans, there are a few yeast autophagy genes missing in the C. elegans genome, including genes encoding critical components of the induction step, (e.g., ATG13, ATG17), or components that act during the vesicle nucleation step (e.g., ATG14). An interesting question is whether functional orthologs of such components exist in C. elegans, but lack apparent structural homology, or whether there is imprecise conservation of the molecular mechanisms of autophagy in the nematode.
Table 1. C. elegans orthologs of S. cerevisiae autophagy-related genes
| S. cerevisiae gene | C. elegans ortholog | Function | Phenotype of Ce gene inactivation | References |
|---|---|---|---|---|
| Regulation of Induction | ||||
| TOR1,2 |
B0261.2 |
PI K-related protein kinase, Rapamycin target | L, LL | |
| ATG1 | unc-51 | Ser/Thr Protein kinase | Unc, Egl, Ad | |
| ATG13 | D2007.5 | Phosphoprotein component of Atg1 complex | Funakoshi et al., 1997 | |
| Vesicle Nucleation | ||||
| ATG6 |
T19E7.4 |
Component of the PI3K complex | L, Ad, St, SL, pQ | |
| VPS34 |
B0025.1 |
PtdIns 3-Kinase | L, SL | |
| VPS15 | ZK930.1 | Component of the PI3K complex | ? | |
| Vesicle Expansion | ||||
| ATG3 | Y55F3AM.4 | E2-like enzyme conjugates PE to Atg8 | ? | |
| ATG4 | ZK792.8 | Cysteine protease cleaves Atg8 C-terminus | ? | Kirisako et al., 2000 |
| Y87G2A.3 | ? | |||
| ATG5 |
Y71G12B.12 |
Conjugated to Atg12 through internal Lys | ? | Kametaka et al., 1996 |
| ATG7 | E1-like enzyme activates Atg8 and Atg12 | SL,Ad, pQ | ||
| ATG8 |
C35D5.9 |
Ubiquitin-like protein conjugated to PE | L, Ad | |
| NE | Meléndez et al., 2003 | |||
| ATG10 | D2085.2 | E2-like enzyme conjugates Atg5 and Atg12 | ? | |
| ATG12 | Ubiquitin-like protein conjugated to Atg5 | SL | ||
| ATG16 | K06A1.5 | Component of Atg5-Atg12 complex | ? | Mizushima, et al., 1999 |
| F02E8.5 | ? | |||
| Recycling | ||||
| ATG2 | M03A8.2 | Peripheral membrane protein interacts with Atg9 | ? | |
| ATG9 |
T22H9.2 |
Integral membrane protein Interacts with Atg2 | NE | Noda et al., 2000 |
| ATG18 |
F41E6.13 |
Peripheral membrane protein binds PI3P and PI(3,5)P | L, Ad, pQ | |
| C. elegans phenotypes are: Unc, uncoordinated; LL, long lifespan; SL, short lifespan; pQ, susceptibility to polyQ expansion disease; Ad, abnormal dauer; Egl, egg laying deficient; St, survival under starvation conditions; L, lethal; NE, no effect;?, not known. | ||||
When comparing the number of autophagy genes in yeast with that of higher eukaryotes, there are multiple orthologs for some autophagy genes. C. elegans and mammals have multiple orthologs of ATG4. In addition, there are two orthologs of ATG8 in the C. elegans genome, although only one may function in autophagy, lgg-1 and not lgg-2 (Meléndez et al., 2003). In mammals, similarly, there are three versions of ATG8 (LC3, GATE-16, and GABARAP), but only LC3 seems to function in autophagy (Kabeya et al., 2000). There also may be some notable functional divergences between yeast and C. elegans autophagy genes, especially for bec-1/ATG6, and unc-51/ATG1. Yeast ATG6 is required for survival during starvation, but not for survival during vegatative growth whereas the C. elegans ortholog, bec-1, is essential for embryonic viability (see section 3.3 below). Moreoever, ATG6 functions in two distinct VPS34 complexes in yeast, one that functions in autophagy and one that functions in vacuolar protein sorting (VPS) (Seaman et al., 1997). However, C. elegans bec-1 can only complement the autophagy but not the VPS function of ATG6 in yeast (Meléndez et al., 2003). Another difference between yeast and mammals is the functional interaction between the antiapoptotic protein Bcl-2 and the autophagy protein Beclin 1 (Maiuri et al., 2007a; Maiuri et al., 2007b; Pattingre et al., 2005). The association of Beclin 1 with Bcl-2 or the Bcl-2 family of antiapoptotic proteins (Bcl -XL, Mcl-1, Bcl-w) inhibits autophagy (Erlich et al., 2007; Maiuri et al., 2007b; Pattingre et al., 2005). When Beclin 1 dissociates from Bcl-2, autophagy is induced. The Bcl-2-Beclin 1 association has been shown to be conserved among the C. elegans orthologs of these proteins, CED-9 and BEC-1 (Takács-Vellai et al., 2005).
The C. elegans ortholog of ATG1, unc-51, (Fig. 2A) appears to have unique functions in the nematode that are distinct from other components of the autophagic machinery. UNC-51 is a neuronal specific protein that functions in axonal elongation and guidance (Hedgecock et al., 1985; Ogura et al., 1994) and the uncordinated phenotype of unc-51 mutation is thought to result from the abnormal localization of UNC-5, the receptor for the axon guidance protein Netrin/UNC-6, in neuronal cell bodies (Ogura and Goshima, 2006). However, RNAi for other autophagy-related genes such as bec-1/ATG6, atg-7/ATG7, lgg-1/ATG8, or atg-18/ATG18, has no effect in the localization of UNC-5 (Ogura and Goshima, 2006), suggesting that the localization of UNC-5 is mediated by an unknown mechanism that does not involve autophagy. Although unc-51 mutants are viable (unlike bec-1 mutants), UNC-51 likely does play a role in autophagy induction similar to its yeast and Drosophila orthologs (Matsuura et al., 1997; Scott et al., 2007) since the phenotype of the unc-51 mutants resembles other autophagy gene mutants, with respect to abnormal dauer development and mislocalization of the autophagy marker, GFP-LGG-1 (Meléndez et al., 2003).
Based on studies in yeast and mammalian cells, it is presumed that, in C. elegans, autophagy occurs at basal levels in most tissues during normal growth conditions, but is rapidly upregulated in response to certain types of environmental stress. This is supported by a low number of LGG-1::GFP punctae in wild-type animals (Hansen et al., 2008; Meléndez et al., 2003). Autophagy can be induced by a number of stimuli, including nutrient starvation, hypoxia, intracellular stress, high temperature, high density, hormones, and various developmental signals (reviewed in Klionsky and Emr, 2000; Levine, 2003; Levine and Klionsky, 2004). Autophagy upregulation occurs when cells need to generate intracellular nutrients and energy (e.g. during starvation, trophic factor withdrawal, or high bioenergetic demands), undergo cellular remodeling (e.g. during developmental transitions), or rid themselves of damaging cytoplasmic components (e.g. during oxidative stress, infection, protein aggregate accumulation, aging). The molecular signaling cascades that regulate autophagy in yeast and mammalian cells have been the subject of recent comprehensive reviews (Levine, 2007; Maiuri et al., 2007c; Meijer and Codogno, 2004; Meijer and Codogno, 2006; Xie and Klionsky, 2007).
Many of the key signaling mechanisms for autophagy regulation in other eukaryotic organisms appear to be conserved in C. elegans. These include the target of rapamycin (TOR, also known as FRAP1), the class I and class III PI3Ks, the insulin/IGF-1-like receptor, and the Ras/Raf/mitogen-activated protein kinase kinase 1/2 (MEK1/2) (Codogno and Meijer, 2005; Meijer and Codogno, 2004). The TOR and Class I PI3Ks pathways negatively regulate autophagy when nutrients and growth factors are abundant, whereas Class III PI3K and MEK1/2 positively regulate autophagy.
In yeast, all ATG genes have been shown to act downstream of TOR (Kamada et al., 2004), however the mechanisms by which nutrients regulate the TOR signaling pathway in yeast and higher eukaryotes, as well as the protein targets of this pathway remain poorly understood. In C. elegans, the TOR ortholog, let-363, encodes, by alternative splicing, two proteins orthologous to S. cerevisiae Tor1 and Tor2, and to human TOR (Long et al., 2002). Loss of let-363 activity, through RNAi or mutation, causes developmental arrest at the L3 stage of larval development, with gonadal and intestinal degeneration and increased hypodermal granules. Furthermore, TOR function affects lifespan and interacts with the DAF-2/insulin-like signaling pathway (Vellai et al., 2003). The let-363 developmental arrest phenotype is phenocopied by RNAi of the orthologs of components of the translation machinery: eIF4G (IFG-1), eIF2α(Y37E3.10), or eIF-2β(IFTB-1), suggesting that this phenotype may arise from failure to promote global translation of mRNA (Long et al., 2002). However, the let-363 phenotype is not phenocopied by the inactivation of C. elegans orthologs of yeast negative regulators of TOR (e.g. SIT4/PPH-4.1 and TIP42/PPFR-3) (Long et al., 2002). Thus, although one has to be careful about interpreting negative RNAi results, it is possible that there are differences in the way that TOR signaling occurs in C. elegans versus S. cerevisiae. An important question regards the extent to which autophagy activation, in the setting of LET-363/TOR inhibition, contributes to the phenotype of let-363 mutants.
The levels of dietary protein or amino acid availability also affect the insulin-like signaling pathway to control metabolism and growth. In C. elegans, the insulin-like growth factor 1 pathway (IGF-1/PI3K) inhibits dauer arrest through the activation of DAF-2, the insulin receptor, AGE-1, a Class I PI3K, and protein kinases PDK-1, AKT-1/ AKT-2, and SGK-1 (Hertweck et al., 2004; Kimura et al., 1997; Morris et al., 1996; Paradis et al., 1999; Paradis and Ruvkun, 1998). Upon activation of the Class I PI3K, the generated PIP3 recruits AKT-1, AKT-2, SGK-1 and PDK-1 kinases to the plasma membrane, where PDK-1 activates AKT and SGK-1 by phosphorylation (Hertweck et al., 2004; Paradis and Ruvkun, 1998). AKT-1, in turn, phosophorylates and inhibits the FOXO transcription factor DAF-16 by promoting its sequestration in the cytoplasm (Hertweck et al., 2004; Lee et al., 2001; Lin et al., 1997; Lin et al., 2001; Ogg et al., 1997). In the absence of ligand, or in the presence of a mutation in any gene upstream (ie., daf-2, age-1, pdk-1, akt-1/2) in the pathway, DAF-16/FOXO is dephosphorylated and translocates into the nucleus (Henderson and Johnson, 2001; Lin et al., 2001). DAF-18, the phosphoinositide 3-phosphatase PTEN, dephosphorylates PI3P and antagonizes AGE-1/PI3K (Gil et al., 1999; Mihaylova et al., 1999; Ogg and Ruvkun, 1998; Rouault et al., 1999). In mammalian cells, the Class I PI3K pathway inhibits autophagy, and this PI3K inhibition is counteracted by the PTEN tumor suppressor (Arico et al., 2001; Petiot et al., 2000). The Class I PI3K regulation of autophagy is conserved in Drosophila (Rusten et al., 2004; Scott et al., 2004) and likely is also conserved in C. elegans, since a loss-of-function mutation in daf-2 increases autophagy (Hansen et al., 2008; Meléndez et al., 2003). Other components of the pathway have not been formally tested yet in the regulation of autophagy in either organism. In the C. elegans genome, there are multiple FOXO orthologs, and it is suspected but unclear if DAF-16 directly or indirectly regulates autophagy (see section 3.4).
In C. elegans, two other pathways regulate dauer development and thus could potentially regulate autophagy: the guanylyl cyclase pathway (DAF-11 is the guanylyl cyclase) and the TGF-β-like pathway (DAF-7 is the ligand (Ren et al., 1996)). These are described in detail in other sections of Wormbook (see TGF-β signaling, or Dauer). daf-7(m62) mutants treated with bec-1 RNAi do not develop into normal dauers when grown at the dauer inducing temperature (Meléndez et al., 2003) (see section 3.2 on the function of autophagy in dauer development), providing some indirect evidence for a role of the TGF-β pathway in controlling autophagy. Although genetic analyses are consistent with DAF-7/TGF-β and DAF-11 pathways functioning in parallel, DAF-11 may also function upstream of TGF-β and IGF-1/PI3K (Thomas et al., 1993). Thus, it is possible that the autophagy pathway is a downstream effector of all three pathways (the IGF-1/PI3K/DAF-2, the TGF-β/DAF-7 and the guanylyl cyclase/DAF-11 pathways); further experiments are necessary to resolve this question. In C. elegans, an additional TGF-β signaling pathway is the Sma/Mab pathway, a major regulator of body size (see TGF-β signaling; Savage et al., 1996). Mutants in this pathway have a small body size phenotype due to reduction in cell size. As of yet, it has not been investigated whether the cause for this smaller cell or body size is an increase in autophagy.
Similar to the insulin-like signaling pathway, the mitogen-activated protein kinase (MAPK) pathway also plays an important role in cellular stress response. The c-Jun N-terminal kinase (JNK) is a member of the MAPK superfamily that is activated by cytokines, including TNF and IL-1, and by exposure to environmental stresses (Kim et al., 2004), and stimulates autophagy in both mammalian cells and Drosophila (Jia et al., 2006; Mills et al., 2004; Pattingre et al., 2009; Wei et al., 2008: Yu et al., 2004). DAF-16/FOXO phosphorylation by JNK, at different sites from AKT phosphorylation, can lead to its enhanced nuclear localization following stress (Oh et al., 2005), indicating that DAF-16 phoshorylation by AKT and JNK may have opposing effects on DAF-16 translocation to the nucleus (and potentially on autophagy regulation). In addition, Greer et al. (2007) have reported that the AMP-activated protein kinase (AMPK), another autophagy-stimulatory kinase in yeast and mammalian cells (for reviews see Høyer-Hansen and Jäättelä, 2007; Meijer and Codogno, 2007), phosphorylates human FOXO3 at an unidentified novel regulatory site. This phosphorylation leads to activation of FOXO3 transcriptional activity without affecting its subcellular localization in mammalian cells. AMPK also phosphorylates DAF-16/FOXO in C. elegans and increases longevity and stress resistance (Curtis et al., 2006). Thus, several different kinases that regulate autophagy in other species can phosphorylate C. elegans DAF-16/FOXO and affect either its localization or its transcriptional activity. It seems likely, but is not yet proven, that these kinases also regulate autophagy in the worm via direct or indirect effects on DAF-16.
The overactivated muscarinic acetylcholine receptor pathway normally responds to starvation in the C. elegans pharynx, and can cause excessive autophagy, pharyngeal muscle dysfunction, and animal death (Kang et al., 2007). Inhibitors of muscarinic signaling such as atropine, a muscarinic acetylcholine receptor antagonist, U0126, an upstream kinase MEK inhibitor, and a reduction-of-function mutation in mpk-1, which encodes a MAPK activated by starvation, all reduce autophagy and the effects of the overactivated muscarinic pathway. Loss of activity of DAPK-1, the C. elegans ortholog of the death-associated protein kinase (DAP kinase), or RGS-2, a regulator of G protein signaling, also reduces starvation-induced autophagy and partially reduces the lethal effects of the overactivated muscarinic acetylcholine pathway. These results suggest that the muscarinic acetylcholine signaling pathway positively regulates autophagy in response to starvation, and that DAPK-1 acts downstream of or in parallel to muscarinic signaling, possibly with RGS-2 in the regulation of autophagy (Kang et al., 2007). These data are consistent with previous reports in mammalian cells (Gozuacik and Kimchi, 2006; Inbal et al., 2002; Pattingre et al., 2003a; Pattingre et al., 2003b; Tallóczy et al., 2002).
In recent studies in mammalian cells four new genes have been identifed that function as positive regulators of autophagy and some have orthologs in C. elegans (Table 2). Three of these genes, UVRAG, Bif-1 and Ambra1, function as inducers of autophagy through their association with Beclin 1, the mammalian ortholog of Atg6/Vps30 (Cecconi et al., 2007; Liang et al., 2006; Takahashi et al., 2007). The fourth gene, DRAM (damage-regulated autophagy modulator), is a p53 target gene encoding a lysosomal protein that induces autophagy (Crighton et al., 2006). UVRAG (UV irradiation resistance-associated gene) encodes a coiled coil protein that positively regulates the Beclin 1-PI3K complex (Liang et al., 2006). UVRAG, like Beclin 1, is a candidate tumor suppressor gene, and is monoallelicaly mutated at a high frequency in certain human cancers (Liang et al., 2006). Bif-1, also known as Endophilin B1, interacts with Beclin 1 through UVRAG, and functions to activate the Beclin 1-PI3K complex and induce autophagosome formation (Takahashi et al., 2007). Ambra1 (activating molecule in Beclin 1-regulated autophagy) is a WD40 protein that acts as a positive regulator of Beclin 1-dependent autophagy (Cecconi et al., 2007). Lack of Ambra 1 activity in mouse embryos leads to severe neural tube defects asssociated with autophagy dysfunction, accumulation of ubiquitinated proteins, uncontrolled cell proliferation, and excessive programmed cell death. Orthologs of Bif-1 and DRAM are found in the C. elegans genome (Table 2), while Ambra1 and UVRAG appear to be missing. It will be interesting to determine whether functional orthologs of UVRAG and Ambra 1 are identified that act in a similar fashion to positively regulate autophagy, although they lack obvious structural homology.
Table 2. C. elegans orthologs of mammalian autophagy-related genes
| Mammalian gene | C. elegans gene | Function | Phenotype of Ce gene inactivation | References |
|---|---|---|---|---|
| UVRAG | - | UV irradiation resistance-associated gene | ? | Liang et al., 2006 |
| Bif-1 | erp-1 | Bax interacting factor that associates with Uvrag and Beclin 1 | ? | Takahashi et al., 2007 |
| DRAM | C33A11.4 | p53-induced damage-regulated | ? | Crighton et al., 2006 |
| Bcl-2 | ced-9 |
Anti-apoptotic protein, negative regulator of Beclin 1-mediated autophagy |
CD | |
| C. elegans phenotypes are: CD, increase in apoptotic cell death; ?, not known. | ||||
To interpret the phenotypes of genetic studies of autophagy in C. elegans, it is useful to have a general understanding of the physiological functions of autophagy. This topic has been reviewed extensively elsewhere (Klionsky, 2007; Levine, 2007; Levine and Kroemer, 2008; Maiuri et al., 2007c; Mizushima, 2007; Mizushima and Klionsky, 2007). Briefly, the major known physiological functions of autophagy in multicellular organisms, as determined largely by knockout studies in mice, include (1) provision of nutrients during catabolism, (2) generation of ATP in stressed or starved cells, (3) generation of signals for heterophagic removal of apoptotic corpses, (4) degradation of misfolded proteins, (5) removal of surplus or damaged organelles, and (6) preservation of genomic stability. The ability of autophagy to defend cells against metabolic stress, through the generation of substrates to maintain macromolecular synthesis and ATP production, likely underlies its evolutionarily conserved role in promoting organismal survival during starvation. The ability of autophagy to function in both routine housekeeping (i.e., basal levels of autophagy are required to prevent the abnormal accumulation of proteins and organelles) as well as in the removal of abnormal or damaged proteins and organelles (e.g., aggregate-prone proteins, damaged mitochondria, peroxisomes, stressed endoplasmic reticulum) likely underlies its roles in preventing tissue degeneration, aging, and potentially, cancer. It is not yet known specifically (or at least not at the level of single cell physiology) whether autophagy performs each of these physiological functions in C. elegans. However, many of the phenotypes of autophagy gene knockout/knockdowns discussed in Section 3 below (e.g., failure to survive during starvation, decreased lifespan extension, susceptibility to polyglutamine expansion-induced disease, etc.) are likely a consequence of autophagy's essential role in nutrient and energy recycling or in protein and organelle breakdown.
Our understanding of the role of autophagy in C. elegans development is emerging from studies using either chromosomal mutations or RNA inactivation of autophagy genes. Chromosomal mutations exist for ATG6/bec-1, TOR/let-363, VPS34/let-512, ATG1/unc-51, and ATG18/atg-18 (Table 1). Inactivation by RNAi is achieved by injection of dsRNA, as was done for bec-1 to uncover its role in longevity and dauer development (Meléndez et al., 2003), as well as RNAi of unc-51, atg-7/M7.5, lgg-1, and atg-18 to uncover the role of autophagy during dauer development (Meléndez et al., 2003). RNAi is also accomplished by feeding worms dsRNA-expressing bacteria, and this has been successfully done for bec-1, atg-7, vps-34 and lgg-3 to show the role of autophagy in lifespan extension (Hansen et al., 2008; Hars et al., 2007; Jia and Levine, 2007; Meléndez et al., 2008), as well as for unc-51, lgg-1, atg-18, and bec-1 to show that autophagy is required for necrotic cell death (Samara et al., 2008; Tóth et al., 2007). Orthologs to yeast ATG3, ATG4, ATG5, ATG10, and ATG16 have still to be tested for a role in autophagy or development (Table 1). C. elegans orthologs to the recently reported mammalian coactivators of the Class III PI3K complex, the UVRAG-associated protein Bif-1, and the p53-induced molecule, DRAM, also exist (Table 2), but have not been tested for their role in autophagy in vivo in C. elegans. Future studies on these genes will likely yield more information on the regulation of autophagy and its role during development.
Prior to the identification of the molecular machinery of autophagy, autophagy was detected primarily by using electron microscopy to visualize autophagosomes. Autophaghosomes are characterized ultrastructurally by the presence of double-membranes surrounding sequestered cytoplasmic material. The electron microscopy approach to detect autophagy is extremely tedious, and difficult to use to assess autophagic activity in different cell types or tissues at the organismal level. Another complicating factor in using electron microscopy to study autophagic activity is the very short half-life of autophagosomes. Indeed, in yeast autophagosomes were originally detected in proteinase-deficient strains, and protease inhibitors are needed to visualize autophagosomes at the microscopic level (Takeshige et al., 1992). In C. elegans subjected to electron microscopic analysis during dauer development, a stage in which autophagic activity is high, it is much easier to detect late structures in the autophagic pathway (e.g., autolysosomes) than intact autophagosomes (Meléndez et al., 2003).
The discovery that one of the autophagy proteins, Atg8 (and its orthologs) undergoes lipidation and stable association with the autophagosomal membrane has facilitated in vivo assays to monitor autophagy. In the absence of autophagy, Atg8 has a diffuse cytoplasmic localization; when autophagy is induced, it tightly associates with the pre-autophagosomal/autophagosomal membrane and has a punctate appearance. Therefore, the monitoring of the intracellular localization of green fluorescent protein-tagged Atg8 molecules has become an effective and reliable method to monitor autophagy in diverse organisms, including C. elegans (Kabeya et al., 2000; Mizushima, 2004; Mizushima et al., 2004).
LGG-1 is the C. elegans ortholog of mammalian LC3 and yeast Atg8, proteins that are commonly used as autophagy markers in yeast and mammalian systems (Huang et al., 2000; Kabeya et al., 2000; Kirisako et al., 1999). To study autophagy in live nematodes, a visual autophagy marker was generated using transgenic worms that express a GFP-tagged LGG-1 protein (Meléndez et al, 2003). In C. elegans, the subcellular pattern of the chimeric GFP-LGG-1 protein (e.g. diffuse versus punctate) can easily be determined under a high-resolution fluorescent microscope. The GFP-LGG-1 molecular marker for autophagy has been validated by its correlation with electron microscopy (Meléndez et al., 2003), and is now considered the method of choice for the detection and quantitation of autophagy in the nematode. Wild-type animals carrying the GFP::LGG-1 extrachromosomal array express the GFP::LGG-1 protein in multiple tissues throughout development, including the nervous system, pharynx, intestine, hypodermis, somatic gonad, and vulva (Meléndez et al., 2003). GFP::LGG-1 has now been used to detect the formation of autophagosomes in several different cell types in C. elegans (ie. this reporter has limited use in some tissues, e.g., intestine in adults), including hypodermal seam cells, intestine, neurons, and muscle (Bamber and Rowland, 2006; Hansen et al., 2008; Kang et al., 2007; Meléndez et al., 2003; Nicot et al., 2006; Tóth et al., 2007). RNAi-mediated knockdown of genes involved in the execution of autophagy result in GFP::LGG-1 forming large aggregates (rather than small punctae that represent “normal” autophaghosomes), presumably due to ineffective clearance of this protein through the autophagolysosomal pathway (Hansen et al., 2008; Meléndez et al., 2003).
Similar to its role in other eukaryotic organisms (Lum et al., 2005; Yorimitsu and Klionsky, 2005), the autophagic machinery is involved in promoting nematode survival during starvation (Kang et al., 2007). Using C. elegans that express the GFP::LGG-1 reporter as an integrated transgene, Kang et al. (2007) found that autophagy is activated by starvation in the pharyngeal muscle. Moreover, an insufficient level of autophagy, caused by either partial RNAi of bec-1 or RNAi against atg-7, promotes the death of animals undergoing starvation. In starved bec-1 RNAi animals, pharyngeal pumping is decreased and can increase with the addition of food. Pumping efficiency correlates with survival, suggesting that autophagy provides the energy essential to maintain pharyngeal pumping efficiency and survival during starvation. It is not yet fully known which other cell types undergo autophagy activation in response to starvation, but GFP::LGG-1 foci have also been observed in seam cells of starved animals (Malene Hansen, personal communication). Presumably, as in mammals, other muscle cells may also be an important site of autophagic catabolism and serve to generate systemic nutrients that can maintain the health of neurons and other vital cells.
While physiological levels of autophagy are adaptive during starvation, Kang et al. (2007) also found that excessive levels of autophagy contribute to nematode death. In gbp-2 (a G protein β subunit involved in RGS-mediated inhibition of the Gqα pathway) mutants, in which muscarinic signaling is hyperactive in the pharyngeal muscle, starvation induces excessive autophagy in pharyngeal muscles, pharyngeal muscle damage, and nematode death that can be partially suppressed by bec-1 or atg-7 RNAi. It is not yet known how excessive autophagy promotes pharyngeal muscle damage or how the damage to the pharyngeal muscle causes death of the animal. Nonetheless, these data provide in vivo genetic evidence that excessive autophagy can promote cellular pathology and organismal death, and underscore the importance of tight regulation of the autophagy pathway so that it can serve adaptive functions (associated with physiological levels) but not maladaptive functions (associated with overactive, non-physiological levels).
The first evidence for a role of autophagy in C. elegans development was provided by studies in daf-2(e1370) mutant dauer animals. During growth in unfavorable environmental conditions (e.g. starvation, high population density, increased temperature) or exposure to “dauer” pheromone, C. elegans larvae arrest in an alternative third larval stage, referred to as the dauer diapause (Cassada and Russell, 1975; Riddle and Albert, 1997). Dauer development is regulated in a temperature-sensitive manner by different signaling pathways that also regulate fat storage, longevity, reproduction and stress responses; including the insulin/IGF-1, the transforming growth factor (TGF-β), and the cyclic guanosine monophosphate (cGMP) pathways (Barbieri et al., 2003; Patterson and Padgett, 2000; Raizen et al., 2006). Dauer larvae undergo a number of metabolic and morphologic changes that permit long-term survival during harsh environmental conditions, such as increased intestinal fat storage, pharyngeal, intestinal and hypodermal constriction, total body elongation, and the formation of a thick cuticle that seals them from the environment. If conditions improve, dauer larvae can resume reproductive development, reach the adult stage, and have a normal lifespan. Although the regulation of dauer development has been extensively characterized, the cellular pathways involved in dauer morphogenesis are less well understood.
Interestingly, the control of dauer development in C. elegans shares similarities with the induction of autophagy in other eukaryotes; environmental cues such as temperature, starvation, and high population, are potent inducers of autophagy in yeast, Dictyostelium and mammals and also induce dauer formation in C. elegans. In dauer larvae with a daf-2 (the insulin-like receptor) mutation, Meléndez et al. (2003) observed an increase in autophagy (detectable both by both visualization of GFP::LGG-1 punctae and by electron microscopy) in the hypodermal seam cells, a cell type required for multiple aspects of dauer morphogenesis. This autophagy induction is required for dauer morphogenesis since the majority of daf-2 animals that are grown at the dauer-restrictive temperature and treated with RNAi against bec-1, unc-51, atg-7, lgg-1, and atg-18 fail to complete normal dauer development (Meléndez et al., 2003). Such animals have less fat storage, do not undergo pharyngeal and total body constriction and elongation, are not resistant to treatment with detergent (sodium dodecyl sulfate or SDS), die within a few days, and fail to resume normal development when tranferred to the non-dauer permissive temperature.
These data suggest that autophagy genes act downstream of DAF-2 insulin/IGF-1 signaling, that autophagy is required for normal dauer morphogenesis, and that autophagy may be essential for the cellular and tissue remodeling that permits C. elegans to successfully adapt to environmental stress. Autophagy is also required for dauer formation triggered by lack of daf-7/TGF-β (Meléndez et al., 2003). The mechanisms by which autophagy is regulated during dauer formation are not known. One possibility is that DAF-16/FOXO, a forkhead transcription factor negatively regulated by insulin-like signaling (Lin et al., 2001), transcriptionally activates components of the autophagic machinery. However, there are no published data to support this from multiple daf-16;daf-2 microarrays experiments. Indeed, several autophagy genes, including lgg-1 and bec-1, contain DAF-16 binding sites; but so do many other genes. In mammals, a constitutively active form of the forkhead transcription factor FOXO3, has been recently shown to induce autophagosome formation in skeletal muscle, as visualized by electron microscopy and GFP-LC3 fluorescence (Mammucari et al., 2007). The transcription of autophagy-related genes such as LC3 (the ATG8/LGG-1 ortholog) and Bnip3 (a BH3-only protein identified as a major effector of FOXO3-mediated autophagy in skeletal muscle) is activated during fasting and is mediated by FOXO3. However, rapamycin fails to induce autophagy in skeletal muscle in vivo; thus it has been hypothesized that an AKT/FOXO3 complex rather than a AKT/TOR complex is involved in the induction of autophagy in skeletal muscle (Mammucari et al., 2008). The complex crosstalk between these pathways and their regulation of autophagy warrants further investigation. Another open question is whether autophagy, in addition to its role in dauer morphogenesis, is also required for dauer survival and/or dauer recovery. One clue suggesting that autophagy may have a role in these processes is the phenotype of nematodes with mutation in pcm-1, a gene that encodes the protein L-isoaspartyl-O-methyltransferase which participates in the repair of age-damaged proteins. pcm-1 mutants display a defect in the ability to recover from starvation-induced L1 larval arrest, and decreased autophagy during dauer formation (Gomez et al., 2007; Gomez and Clarke, 2007). Thus, the lack of normal protein repair may interfere with the autophagic pathway, although it is not yet known whether the defect in autophagy is mechanistically responsible for the survival and recovery defects of pcm-1 mutants.
The role of autophagy in early development in C. elegans is not confined to developmental changes that occur in adaptation to stress. High concentration of dsRNA directed against C. elegans orthologs of ATG6 (bec-1), ATG8 (lgg-1), and ATG18 (F41E6.13) result in early developmental arrest, during or before the first larval stage (Meléndez et al., 2003). A deletion allele of atg-18 is available from the C. elegans Gene Knockout Consortium, and described as superficially wild-type in WormBase. It is possible that the deletion does not completely inactivate the gene or that the atg-18 RNAi effect was not due to the inactivation of atg-18 alone. Nevertheless, worms carrying a mutation in the BEC-1 partner protein, the Class III PI3K Vps34 (C. elegans ortholog is let-512), also display a lethal phenotype (Roggo et al., 2002). In addition, bec-1 homozygous animals derived from a bec-1 heterozygous parent arrest at various stages of development and exhibit increased vacuolization, uncoordinated phenotypes, and molting defects (Takács-Vellai et al., 2005). Mutant bec-1 animals that reach adulthood are sterile. bec-1 mutants also display a maternal effect lethality; animals that lack both the maternal and zygotic expression of bec-1 die early during embryogenesis (unpublished, A.M.). Thus, bec-1 activity is required for viability and fertility (Takács-Vellai et al., 2005). It is not yet known whether other autophagy gene mutations confer similar phenotypes, nor specifically how the lack of BEC-1 function (and presumably autophagy) results in these developmental defects.
Multiple processes, such as neuroendocrine signaling, mitochondrial function, and nutritional sensing, play a role in determining lifespan in C. elegans. Experiments in C. elegans have provided direct evidence for a role of autophagy in lifespan extension in at least two settings: mutations in the insulin-like signaling pathway and dietary restriction (Hars et al., 2007; Jia and Levine, 2007; Meléndez et al., 2003). In C. elegans, mutations in the endocrine signaling pathway, involving the insulin-like growth factor (IGF-1) receptor DAF-2 and the PI3K AGE-1, display an increase in adult longevity that requires the forkhead transcription factor FOXO/DAF-16 (reviewed in Kenyon, 2005). Meléndez et al. (2003) found that autophagy inactivation by RNAi of bec-1, decreases lifespan of daf-2 animals at doses that have minimal or no effect on the lifespan of N2 animals. In addition, Hars et al. (2007) showed that RNAi of the atg-7 and atg-12 C. elegans orthologs shortened the lifespan of both N2 (wild-type) and daf-2 mutants, but the decrease in lifespan was greater in the daf-2 mutants. Thus, multiple autophagy genes are required for lifespan extension in daf-2 (IGF-1) mutants.
Dietary restriction plays an evolutionarily conserved role in lifespan extension in yeasts, flies, mammals, and C. elegans. The correlation between increased autophagy and increased lifespan in feeding-defective C. elegans mutants, eat-2, eat-3 and pha-3, provided a clue that autophagy might be involved in dietary restriction-mediated lifespan extension (Curtis et al., 2006; Kaeberlein et al., 2006; Lee et al., 2006; Mörck and Pilon, 2006). Mutations in TOR, a nutrient sensor downstream of the dietary-restriction response that negatively regulates autophagy, also extend lifespan (Hansen et al., 2007; Vellai et al., 2003). Recently, the autophagy genes, bec-1, vps-34 and atg-7, have been shown to be required for the lifespan extension of eat-2 mutants (Hansen et al., 2008; Jia and Levine, 2007). In addition, the specific inhibition of autophagy during adulthood shortens the long lifespan of eat-2 and TOR pathway mutants (Hansen et al., 2008; Meléndez et al., 2008) indicating that the autophagic machinery is indeed part of the mechanism by which TOR inhibition increases lifespan, and supporting the idea that autophagy is required for dietary restriction-mediated lifespan extension (Hansen et al., 2008; Jia and Levine, 2007). The specific inhibition of autophagy genes by RNAi treatment during adulthood eliminates the potential complication of autophagy having a developmental role that affects longevity. Since autophagy and longevity regulation are highly conserved from C. elegans to mammals, a similar role for autophagy in dietary restriction-mediated lifespan extension may also exist in mammals.
The mechanisms by which autophagy mediates lifespan extension are not yet understood. However, one possibility is that the increase in lifespan is mediated through the autophagy-dependent nonspecific or selective removal of damaged mitochondria, decrease in levels of intracellular reactive oxygen species, and subsequent protection against oxidative damage. Such a mechanism would overlap with the presumed means by which autophagy may protect against genomic instability and tumor progression. It would also provide a framework for the conceptual integration of the oxidative damage theory of aging and the known stimuli that influence aging through autophagy-dependent mechanisms in C. elegans, including dietary restriction and insulin-like signaling. Many of the long-lived mutants in C. elegans are resistant to oxidative stress and many mutations that decrease mitochondrial electron transport are long-lived, whereas conversely, mutations that increase oxidative damage shorten lifespan in C. elegans (Antebi, 2007; Guarente and Kenyon, 2000). Thus, longevity in C. elegans (and potentially other organisms) may be mediated either by mutations that directly affect cellular generation or breakdown of reactive oxygen species, or indirectly, decrease reactive oxygen specifies via upregulation of the autophagic turnover of the damaged organelles that generate these harmful species.
As with autophagy induction during dauer diapause, it is not yet known which transcription factors regulate autophagy in long-lived mutant nematodes. Unlike the DAF-2/insulin/IGF-1 signaling pathway, dietary restriction is not dependent on the FOXO transcription factor DAF-16 (Houthoofd et al., 2005; Lakowski and Hekimi, 1998). Instead, the FOXA transcription factor PHA-4 is required for the longevity of dietary-restricted mutants (Brenkman and Burgering, 2007; Panowski et al., 2007), as pha-4 mutants are able to completely suppress the long lifespan of eat-2 mutant animals. Hansen et al. (2008) have shown that pha-4 is required for eat-2 mutants to have elevated numbers of GFP::LGG-1 foci. Reduced daf-2/insulin/IGF-1 signaling mutants lacking daf-16/FOXO activity still show high levels of autophagic vesicles as measured by the LGG-1::GFP reporter, even though the daf-16; daf-2 double mutant animals are short-lived (Hansen et al., 2008). Thus, an increase in the levels of autophagy does not appear to be sufficient to extend lifespan in the context of reduced daf-2/insulin IGF-1 signaling. One interesting point is that overexpression of pha-4 extends longevity only in the absence of daf-16 activity (Panowski et al., 2007), suggesting that the role of pha-4 and daf-16 may be partially redundant with regards to longevity or that there is inherent competition for these two transcription factors. How this may relate to autophagy remains unclear.
Inactivation of several mitochondrial electron transport (ETC) genes in C. elegans also extends lifespan (Dillin et al., 2002; Lee et al., 2003). Whether autophagy is specifically involved in mitochondrial-mediated lifespan extension is not clear. In order to observe a longevity phenotype, RNAi treatment of ETC genes has to be initiated during development, specifically in the L3/L4 stage (Dillin et al., 2002; Rea et al., 2007). Inhibiting the ETC genes by RNAi only during adulthood has no effect on lifespan (Dillin et al., 2002). In long-lived mitochondrial mutants, inactivation of autophagy genes by RNAi delivered throughout the adult life had no effect (Hansen et al., 2008). In contrast, the ETC RNAi treatment delivered throughout development cannot fully extend the lifespan of mutants defective in autophagy, suggesting that autophagy is required for the longevity of mutants with reduced mitochondrial respiration (Tóth et al., 2008). However, the short lifespan of autophagy defective mutants may be partly or fully due to developmental defects. Thus, it remains to be experimentally tested whether autophagy genes are specifically involved in the lifespan extension of mitochondrial mutants that have been RNAi-treated specifically in the L3/L4 stage.
Autophagy has been historically classified as type II programmed cell death based on the presence of autophagic vacuoles in dying cells. However, the question of whether autophagy is a death mechanism per se has been an area of intense debate. In Drosophila, the developmental process of insect metamorphosis involves the massive destruction and elimination of larval tissues, such as the salivary glands and larval gut, through a dramatic upregulation of autophagy (Baehrecke, 2005). However, components of the apoptotic machinery are also upregulated during metamorphosis and seem to act cooperatively with autophagy for the efficient elimination of larval tissues (Berry and Baehrecke, 2007; Juhász et al., 2007; Muro et al., 2006).
The well-recognized role of autophagy as a survival mechanism during stress seemingly contradicts a role in cell death. When autophagosomes are present in dying cells, it is often unclear if autophagy represents a form of nonapoptotic cell death or a failed strategy of cell survival (reviewed in Levine and Yuan, 2005; Maiuri et al., 2007c). A third possibility, suggested by recent studies in mammalian autophagy-defective embryoid bodies (Qu et al., 2007), is that autophagy is activated in dying cells to generate energy-dependent engulfment signals and successful apoptotic corpse clearance.
In mammalian cells, most studies indicating that autophagy can be involved in the death execution process are in cells that have defects in apoptosis (Levine and Yuan, 2005; Maiuri et al., 2007c). As of yet, there is no evidence that autophagy functions as a death mechanism in physiological settings in cells with an intact apopototic pathway or in vivo during developmental cell death. In fact, most autophagy gene knockout organisms, including in C. elegans, display increased rather than decreased cell death, supporting a pro-survival role for autophagy during development. Studies of C. elegans bec-1 null mutants, carrying the chromosomal deletion ok691, support the concept that autophagy plays a pro-survival role at the cellular and organismal level during development. These animals have a highly penetrant lethal phenotype during normal growth conditions, and have been reported to display an early arrest phenotype with an increase in CED-3/caspase dependent apoptotic cell death (Takács-Vellai et al., 2005). While these results suggest that lack of autophagy may trigger the apoptotic pathway, it is also possible that, similar to mammalian development (Qu et al., 2007), the observed increase in apoptotic corpses in bec-1 mutant embryos may represent a defect in apoptotic corpse clearance. The cellular focus for the bec-1 lethal phenotype, and the question of whether autophagy functions in a cell autonomous fashion in development remains unknown.
Parallel to findings in mammalian cells, it is likely that cross-talk occurs between the apoptotic and autophagic machinery in C. elegans. For example, the interaction between the mammalian anti-apoptotic protein Bcl-2 and autophagy protein Beclin 1 (Maiuri et al., 2007b; Pattingre et al., 2005) is conserved among the C. elegans orthologs of these proteins, CED-9 and BEC-1 (Takács-Vellai et al., 2005) (Fig. 2B) Tacaks-Vellai et al. (2005) found that the C. elegans orthologs of these proteins, CED-9 and BEC-1, interact. It is not yet known, however, whether CED-9, like Bcl-2, functions as an anti-autophagy protein. Yet, recent studies with the BH3-only protein, EGL-1, from C. elegans hint that this may be the case. The interaction between mammalian Bcl-2/Bcl-xL involves a BH3 domain within Beclin 1, and this interaction can be competitively disrupted by overexpression of BH3-only pro-apoptotic proteins or pharmacological BH3 mimetics (Maiuri et al., 2007a; Maiuri et al., 2007b). Similarly, a gain-of-function mutation of egl-1 induces autophagy, while deletion of egl-1 compromises starvation-induced autophagy (Maiuri et al., 2007b). This suggests that EGL-1 may not only positively regulate programmed cell death by interacting with CED-9 to induce CED-4 release from CED-4/CED-9 complexes, but also may positively regulate autophagy by interacting with CED-9 to induce BEC-1 release from CED-9/BEC-1 complexes. Whether this increase in autophagy in egl-1 mutants contributes to cell death is not yet clear. Future studies in C. elegans should further clarify the molecular cross-talk between the autophagic and apoptotic machinery during development.
Another open question is whether autophagy plays a role in necrotic cell death. In C. elegans, gain-of-function mutations in ion channel genes, either mec-4 or deg-1, the acetylcholine receptor channel subunit gene, deg-3, and the Gs protein α-subunit gene, gsa-1, cause a necrotic-like cell degeneration in neurons that express the mutant proteins (Berger et al., 1998; Chalfie and Wolinsky, 1990; Driscoll and Chalfie, 1991; Korswagen et al., 1997; Treinin and Chalfie, 1995). Electron microscopy studies of dying neurons in animals carrying the gain-of-function mec-4(d) allele, have shown extensive degradation of cellular contents during the process of necrosis (Hall et al., 1997). This ultrastructural feature is reminiscent of autophagy and does not require the caspase CED-3, which mediates programmed cell-death, nor CSP-1, CSP-2 or CSP-3 (CED-3 related proteases)(Chung et al., 2000), indicating that a distinct, non-apoptotic mechanism may function in neurodegeneration (Syntichaki et al., 2002). Inactivation of three autophagy genes (unc-51, bec-1, and lgg-1) suppresses the degeneration of neurons with hyperactive toxic ion channels (Tóth et al., 2007). These observations indicate a possible role for autophagy in cellular necrosis, and suggest that autophagy inhibition may protect neurons against necrotic cell death. However, inactivation of the TOR-kinase-mediated signaling pathway, a negative regulator of autophagy, also has a protective function in the same neurons (Tóth et al., 2007). These data are difficult to interpret, since inactivation of TOR, a negative regulator of autophagy, should increase autophagy and thus should not have the same effects as autophagy gene inactivation. More recently, Samara et al. (2008) also showed that excessive autophagosome formation is induced early during necrotic cell death and that the autophagy pathway synergizes with lysosomal proteolytic mechanisms to facilitate necrotic cell death in neurons.
The autophagy pathway may play an important role in the clearance of mutant aggregate-prone proteins associated with chronic human neurodegenerative diseases (e.g., Huntington's disease, Parkinson's disease, Alzheimer's disease), muscle diseases (e.g., sporadic inclusion body myositis, limb-girdle muscular dystrophy type 2B, Miyoshi myopathy), and liver disease (e.g., α1-antitrypsin deficiency). Until recently, evidence in support of this stemmed primarily from pharmacological studies in mice and Drosophila using regulators of autophagy such as the TOR inhibitor, rapamcyin, that have pleiotropic effects on the cell. Recent studies in C. elegans provide more direct genetic evidence that the autophagic machinery protects against diseases caused by aggregate-prone proteins associated with human neurodegenerative and muscular diseases.
There are two established models of polyglutamine (polyQ) expansion protein-induced disease in C. elegans. One model involves the expression of fluorescent-tagged proteins containing polyQ expansion tracts in muscle cells. Transgenic animals that express polyQ fragments containing 40 repeats (Q40-YFP) display visible aggregates in muscle cells and impaired locomotion (Brignull et al., 2006; Morley et al., 2002). A second model utilizes the expression of human huntingtin fragments contaning a tract of 150 polyQ residues (Htn-150) in ASH sensory neurons (Jia et al., 2007; Voisine et al., 2007). These animals accumulate Htn-150 aggregates in the ASH neurons and undergo progressive neuronal degeneration. In both models, RNAi against the autophagy genes, bec-1, atg-7, atg-18, increases the accumulation of disease protein aggregates and enhances their toxicity (Jia et al., 2007), indicating a role for autophagy genes in vivo in the suppression of polyQ aggregate accumulation and protection against disease caused by polyQ toxicity.
In another C. elegans model, transgenic animals express human β-amyloid peptide in muscle, providing an opportunity to examine the mechanism of toxicity of aggregate-prone Aβ peptides (Florez-McClure et al., 2007). In this nematode model, Aβ expression inhibits the maturation of autophagosomes into degradative autolysosomes, resulting in the abnormal accumulation of autophagosomes (and presumably blockade of the neuroprotective effects of successful autophagy); this pathology parallels that observed in the brains of patients with Alzheimer's disease and the muscles of patients with sporadic inclusion body myositis (Askanas and Engel, 2006; Murphy and Golde, 2006). The autophagy genes bec-1 and atg-7 appear to be required for the degradation of Aβ in the C. elegans model (Florez-McClure et al., 2007). These observations in C. elegans support the concept that pharmacological activation of the autophagy pathway may be beneficial in the treatment of Alzheimer's disease and other human neurodegenerative disorders.
Similar to reports in Drosophila (Neufeld, 2007; Scott et al., 2007), the activation of autophagy in C. elegans may be associated with a reduction in cell size. Mörck and Pilon (2006) examined the level of autophagy, fat deposits, and body and cell size in feeding-defective mutants like pha-2 and pha-3, which have abnormal pharyngeal anatomy; eat-1, eat-2 and eat-3 mutants, which have reduced pharyngeal pumping rates; and eat-10 mutants, which have a slippery pharynx that inefficiently traps bacteria. All of these feeding-defective mutants are short and exhibit a reduced body width and cell size (most notable during the L4 stage), independently of the nature of the genetic defect that impairs their feeding behavior. This reduction in cell and body size is associated with an upregulation of autophagy, as observed by analyzing the subcellular localization of the GFP::LGG-1 autophagy marker in the hypodermal seam cells of L3 animals. Whether the increase in autophagy directly decreases the cell size of the feeding-defective mutants is not yet known.
In a recent article by Aladzsity et al. (2007), inactivation of bec-1 and unc-51 results in a small cell and body size phenotype; however, whether this is due to a lack of autophagy is not shown. The authors suggests that both excessive and insufficient levels of autophagy lead to retarded cell growth (i.e. small cell and body size), but a mechanism by which both an excess and lack of autophagy would result in a similar phenotype is not provided. In Drosophila, conditions that stimulate autophagy, such as starvation, have been shown to result in reduced cell growth (Neufeld, 2003).
A novel function for autophagy in GABAA receptor degradative trafficking has been shown in C. elegans (Rowland et al., 2006). The clustering of neurotransmitter receptors is the end result of a developmental pathway that is initiated when presynaptic terminals contact the postsynaptic cell. In C. elegans, body-wall muscles form synapses with both GABA and non-GABA neurons (White et al., 1986). GABA terminals organize GABAA receptors into synaptic clusters and in the absence of presynaptic inputs, GABAA receptors are internalized and degraded through autophagy. In these neurons, autophagy is used selectively to degrade GABAA receptors, since acetylcholine receptors in the same cells do not traffic to autophagosomes (Rowland et al., 2006). These findings demonstrate a novel role for autophagy in the degradation of cell surface receptors. In addition to a possible function in controlling the balance of neuronal excitation and inhibition (due to selective GABAA receptor degradation), the selective degradation of other cell surface receptors by autophagy may help regulate cellular growth, differentiation, and development.
Our understanding of the role of autophagy as an essential process in C. elegans development has advanced greatly in the past few years. We have learned that autophagy is involved in cell survival, cell death, normal development, dauer development, negative control of cell growth, longevity, clearance of toxic aggregate-prone proteins, and cell surface receptor trafficking. Despite the great importance of autophagy in the physiology of higher eukaryotes, the molecular mechanisms and functions of autophagy in these organisms remain vague. Hopefully, C. elegans, as a genetically tractable system, will continue to allow us to unravel the mechanisms of autophagy regulation, execution, and function at the cellular and organismal level.
The work in the authors' laboratories is supported by grants from the National Institutes of Health (A.M., B.L.), the American Cancer Society (B.L.), the National Science Foundation (A.M.), the PSC CUNY Research Award Program (A.M.) and the Ellison Medical Foundation (B.L., A.M.). We thank Hannes Bülow and Malene Hansen for critical reading and helpful suggestions, and Renee Talley for administrative assistance.
Aladzsity, I., Tóth, M. L., Sigmond, T., Szabó, E., Bicsák, B., Barna, J., Regos, A., Orosz, L., Kovács, A. L., and Vellai, T. (2007). Autophagy genes unc-51 and bec-1 are required for normal cell size in Caenorhabditis elegans. Genetics 177, 655-660. Abstract Article
Antebi, A. (2007). Genetics of aging in Caenorhabditis elegans. PLoS Genet. 3, 1565-1571. Abstract Article
Arico, S., Petiot, A., Bauvy, C., Dubbelhuis, P. F., Meijer, A. J., Codogno, P., and Ogier-Denis, E. (2001). The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol.Chem. 276, 35243-35246. Abstract Article
Askanas, V., and Engel, W. K. (2006). Inclusion-body myositis: a myodegenerative conformational disorder associated with Ab, protein misfolding, and proteasome inhibition. Neurology 66, S39-48. Abstract
Baehrecke, E. H. (2005). Autophagy: dual roles in life and death? Nat. Rev. Mol. Cell Biol. 6, 505-510. Abstract Article
Bamber, B. A., and Rowland, A. M. (2006). Shaping cellular form and function by autophagy. Autophagy 2, 247-249. Abstract
Barbieri M, Franceschi C, Paolisso, G. (2003). Insulin/IGF-1-signaling pathway: An evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 285, E1064-1071. Abstract
Barth, H., Meiling-Wesse, K., Epple, U. D., and Thumm, M. (2001). Autophagy and the cytoplasm to vacuole targeting pathway both require Aut10p. FEBS Lett. 508, 23-28. Abstract Article
Berg, T. O., Fengsrud, M., Strømhaug, P. E., Berg, T., and Seglen, P. O. (1998). Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J. Biol. Chem. 273, 21883-21892. Abstract Article
Berger, A. J., Hart, A. C., and Kaplan, J. M. (1998). Gαs-induced neurodegeneration in Caenorhabditis elegans. J. Neurosci. 18, 2871-2880. Abstract
Berry, D. L., and Baehrecke, E. H. (2007). Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 131, 1137-1148. Abstract Article
Brenkman, A. B., and Burgering, B. M. (2007). Live longer through PHAsting. Cell Metab. 5, 407-409. Abstract Article
Brignull, H. R., Morley, J. F., Garcia, S. M., and Morimoto, R. I. (2006). Modeling polyglutamine pathogenesis in C. elegans. Methods Enzymol. 412, 256-282. Abstract Article
Cassada, R. C., and Russell, R. L. (1975). The dauer larva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev. Biol. 46, 326-342. Abstract Article
Cecconi, F., Di Bartolomeo, S., Nardacci, R., Fuoco, C., Corazzari, M., Giunta, L., Romagnoli, A., Stoykova, A., Chowdhury, K., Fimia, G. M., and Piacentini, M. (2007). A novel role for autophagy in neurodevelopment. Autophagy 3, 506-508. Abstract
Chalfie, M., and Wolinsky, E. (1990). The identification and suppression of inherited neurodegeneration in Caenorhabditis elegans. Nature 345, 410-416. Abstract Article
Chung, S., Gumienny, T. L., Hengartner, M. O., and Driscoll, M. (2000). A common set of engulfment genes mediates removal of both apoptotic and necrotic cell corpses in C. elegans. Nat. Cell Biol. 2, 931-937. Abstract Article
Codogno, P., and Meijer, A. J. (2005). Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12 Suppl 2, 1509-1518. Abstract
Crighton, D., Wilkinson, S., O'Prey, J., Syed, N., Smith, P., Harrison, P. R., Gasco, M., Garrone, O., Crook, T., and Ryan, K. M. (2006). DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121-134. Abstract Article
Curtis, R., O'Connor, G., and DiStefano, P. S. (2006). Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell 5, 119-126. Abstract Article
Dillin, A., Hsu, A. L., Arantes-Oliveira, N., Lehrer-Graiwer, J., Hsin, H., Fraser, A. G., Kamath, R. S., Ahringer, J., and Kenyon, C. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398-2401. Abstract Article
Driscoll, M., and Chalfie, M. (1991). The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degeneration. Nature 349, 588-593. Abstract Article
Dunn, W. A., Jr. (1994). Autophagy and related mechanisms of lysosome-mediated protein degradation. Trends Cell Biol. 4, 139-143. Abstract Article
Erlich, S., Mizrachy, L., Segev, O., Lindenboim, L., Zmira, O., Adi-Harel, S., Hirsch, J. A., Stein, R., and Pinkas-Kramarski, R. (2007). Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy 3, 561-568. Abstract
Florez-McClure, M. L., Hohsfield, L. A., Fonte, G., Bealor, M. T., and Link, C. D. (2007). Decreased insulin-receptor signaling promotes the autophagic degradation of β-amyloid peptide in C. elegans. Autophagy 3, 569-580. Abstract
Funakoshi, T., Matsuura, A., Noda, T., and Ohsumi, Y. (1997). Analyses of APG13 gene involved in autophagy in yeast, Saccharomyces cerevisiae. Gene 192, 207-213. Abstract Article
Gil, E. B., Malone Link, E., Liu, L. X., Johnson, C. D., and Lees, J. A. (1999). Regulation of the insulin-like developmental pathway of Caenorhabditis elegans by a homolog of the PTEN tumor suppressor gene. Proc. Natl. Acad. Sci. USA 96, 2925-2930. Abstract Article
Gomez, T. A., Banfield, K. L., Trogler, D. M., and Clarke, S. G. (2007). The L-isoaspartyl-O-methyltransferase in Caenorhabditis elegans larval longevity and autophagy. Dev. Biol. 303, 493-500. Abstract Article
Gomez, T. A., and Clarke, S. G. (2007). Autophagy and insulin/TOR signaling in Caenorhabditis elegans pcm-1 protein repair mutants. Autophagy 3, 357-359. Abstract
Gonzalez-Polo, R. A., Carvalho, G., Braun, T., Decaudin, D., Fabre, C., Larochette, N., Perfettini, J. L., Djavaheri-Mergny, M., Youlyouz-Marfak, I., Codogno, P., et al. (2005). PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor. Oncogene 24, 7503-7513. Abstract Article
Gozuacik, D., and Kimchi, A. (2006). DAPk protein family and cancer. Autophagy 2, 74-79. Abstract
Greer, E. L., Oskoui, P. R., Banko, M. R., Maniar, J. M., Gygi, M. P., Gygi, S. P., and Brunet, A. (2007). The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J. Biol. Chem. 282, 30107-30119. Abstract Article
Guarente, L., and Kenyon, C. (2000). Genetic pathways that regulate ageing in model organisms. Nature 408, 255-262. Abstract Article
Hall, D. H., Gu, G., Garcia-Añoveros, J., Gong, L., Chalfie, M., and Driscoll, M. (1997). Neuropathology of degenerative cell death in Caenorhabditis elegans. J. Neurosci. 17, 1033-1045. Abstract
Hansen, M., Chandra, A., Mitic, L. L., Onken, B., Driscoll, M., and Kenyon, C. (2008). A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4, e24. Abstract
Hansen, M., Taubert, S., Crawford, D., Libina, N., Lee, S. J., and Kenyon, C. (2007). Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell 6, 95-110. Abstract Article
Hars, E. S., Qi, H., Ryazanov, A. G., Jin, S., Cai, L., Hu, C., and Liu, L. F. (2007). Autophagy regulates ageing in C. elegans. Autophagy 3, 93-95. Abstract
Hedgecock, E. M., Culotti, J. G., Thomson, J. N., and Perkins, L. A. (1985). Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev. Biol. 111, 158-170. Abstract Article
Henderson, S. T., and Johnson, T. E. (2001). daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 11, 1975-1980. Abstract Article
Hengartner, M. O., and Horvitz, H. R. (1994). C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 76, 665-676. Abstract Article
Hertweck, M., Gobel, C., and Baumeister, R. (2004). C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev. Cell 6, 577-588. Abstract Article
Houthoofd, K., Johnson, T. E., and Vanfleteren, J. R. (2005). Dietary restriction in the nematode Caenorhabditis elegans. J. Gerontol. A Biol. Sci. Med. Sci. 60, 1125-1131. Abstract
Høyer-Hansen, M., and Jäättelä, M. (2007). AMP-activated protein kinase: a universal regulator of autophagy? Autophagy 3, 381-383. Abstract
Huang, W. P., Scott, S. V., Kim, J., and Klionsky, D. J. (2000). The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J. Biol. Chem. 275, 5845-5851. Abstract Article
Ichimura, Y., Kirisako, T., Takao, T., Satomi, Y., Shimonishi, Y., Ishihara, N., Mizushima, N., Tanida, I., Kominami, E., Ohsumi, M., et al. (2000). A ubiquitin-like system mediates protein lipidation. Nature 408, 488-492. Abstract
Inbal, B., Bialik, S., Sabanay, I., Shani, G., and Kimchi, A. (2002). DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157, 455-468. Abstract Article
Jia, G., Cheng, G., Gangahar, D. M., and Agrawal, D. K. (2006). Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol. Cell Biol. 84, 448-454. Abstract Article
Jia, K., Chen, D., and Riddle, D. L. (2004). The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131, 3897-3906. Abstract Article
Jia, K., Hart, A. C., and Levine, B. (2007). Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3, 21-25. Abstract
Jia, K., and Levine, B. (2007). Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy 3, 597-599. Abstract
Juhász, G., Erdi, B., Sass, M., and Neufeld, T. P. (2007). Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 21, 3061-3066. Abstract Article
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., Kominami, E., Ohsumi, Y., and Yoshimori, T. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720-5728. Abstract Article
Kaeberlein, T. L., Smith, E. D., Tsuchiya, M., Welton, K. L., Thomas, J. H., Fields, S., Kennedy, B. K., and Kaeberlein, M. (2006). Lifespan extension in Caenorhabditis elegans by complete removal of food. Aging Cell 5, 487-494. Abstract Article
Kamada, Y., Sekito, T., and Ohsumi, Y. (2004). Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr. Top. Microbiol. Immunol. 279, 73-84. Abstract
Kametaka, S., Matsuura, A., Wada, Y., and Ohsumi, Y. (1996). Structural and functional analyses of APG5, a gene involved in autophagy in yeast. Gene 178, 139-143. Abstract Article
Kametaka, S., Okano, T., Ohsumi, M., and Ohsumi, Y. (1998). Apg14p and Apg6/Vps30p form a protein complex essential for autophagy in the yeast, Saccharomyces cerevisiae. J. Biol. Chem. 273, 22284-22291. Abstract Article
Kang, C., You, Y., and Avery, L. (2007). Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev. 21, 2161-2171. Abstract Article
Kenyon, C. (2005). The plasticity of aging: insights from long-lived mutants. Cell 120, 449-460. Abstract Article
Kihara, A., Noda, T., Ishihara, N., and Ohsumi, Y. (2001). Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 152, 519-530. Abstract Article
Kim, D. H., Liberati, N. T., Mizuno, T., Inoue, H., Hisamoto, N., Matsumoto, K., and Ausubel, F. M. (2004). Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc. Natl. Acad. Sci. USA 101, 10990-10994. Abstract Article
Kim, J., Dalton, V. M., Eggerton, K. P., Scott, S. V., and Klionsky, D. J. (1999). Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol. Biol. Cell 10, 1337-1351. Abstract
Kim, J., Huang, W. P., Strømhaug, P. E., and Klionsky, D. J. (2002). Convergence of multiple autophagy and cytoplasm to vacuole targeting components to a perivacuolar membrane compartment prior to de novo vesicle formation. J. Biol. Chem. 277, 763-773. Abstract Article
Kimura, K. D., Tissenbaum, H. A., Liu, Y., and Ruvkun, G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942-946. Abstract Article
Kimura, S., Noda, T., and Yoshimori, T. (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452-460. Abstract
Kirisako, T., Baba, M., Ishihara, N., Miyazawa, K., Ohsumi, M., Yoshimori, T., Noda, T., and Ohsumi, Y. (1999). Formation process of autophagosome is traced with Apg8/Aut7p in yeast. J. Cell Biol. 147, 435-446. Abstract Article
Kirisako, T., Ichimura, Y., Okada, H., Kabeya, Y., Mizushima, N., Yoshimori, T., Ohsumi, M., Takao, T., Noda, T., and Ohsumi, Y. (2000). The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 151, 263-276. Abstract Article
Klionsky, D. J. (2007). Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931-937. Abstract Article
Klionsky, D. J., and Emr, S. D. (2000). Autophagy as a regulated pathway of cellular degradation. Science 290, 1717-1721. Abstract Article
Korswagen, H. C., Park, J. H., Ohshima, Y., and Plasterk, R. H. (1997). An activating mutation in a Caenorhabditis elegans Gs protein induces neural degeneration. Genes Dev. 11, 1493-1503. Abstract Article
Kovács, A. L., Vellai, T. and F. Müller (2004). Autophagy in Caenorhabditis elegans. In Autophagy, D. Klionsky, ed. (Austin, TX, Landes Bioscience), pp. 219-225.
Lakowski, B., and Hekimi, S. (1998). The genetics of caloric restriction in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 95, 13091-13096. Abstract Article
Lee, G. D., Wilson, M. A., Zhu, M., Wolkow, C. A., de Cabo, R., Ingram, D. K., and Zou, S. (2006). Dietary deprivation extends lifespan in Caenorhabditis elegans. Aging Cell 5, 515-524. Abstract Article
Lee, R. Y., Hench, J., and Ruvkun, G. (2001). Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr. Biol. 11, 1950-1957. Abstract Article
Lee, S. S., Lee, R. Y., Fraser, A. G., Kamath, R. S., Ahringer, J., and Ruvkun, G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40-48. Abstract Article
Legakis, J. E., Yen, W. L., and Klionsky, D. J. (2007). A cycling protein complex required for selective autophagy. Autophagy 3, 422-432. Abstract
Levine, B. (2003). Autophagy in development, tumor suppression, and innate immunity. Harvey Lect. 99, 47-76. Abstract
Levine, B., and Klionsky, D. J. (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463-477. Abstract Article
Levine, B., and Kroemer, G. (2008). Autophagy in the pathogenesis of disease. Cell 132, 27-42. Abstract Article
Levine, B., and Yuan, J. (2005). Autophagy in cell death: an innocent convict? J. Clin. Invest. 115, 2679-2688. Abstract Article
Liang, C., Feng, P., Ku, B., Dotan, I., Canaani, D., Oh, B. H., and Jung, J. U. (2006). Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat. Cell Biol. 8, 688-699. Abstract Article
Liang, X. H., Jackson, S., Seaman, M., Brown, K., Kempkes, B., Hibshoosh, H., and Levine, B. (1999). Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672-676. Abstract Article
Lin, K., Dorman, J. B., Rodan, A., and Kenyon, C. (1997). daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278, 1319-1322. Abstract Article
Lin, K., Hsin, H., Libina, N., and Kenyon, C. (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet. 28, 139-145. Abstract Article
Long, X., Spycher, C., Han, Z. S., Rose, A. M., Müller, F., and Avruch, J. (2002). TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr. Biol. 12, 1448-1461. Abstract Article
Lum, J. J., DeBerardinis, R. J., and Thompson, C. B. (2005). Autophagy in metazoans: cell survival in the land of plenty. Nat. Rev. Mol. Cell Biol. 6, 439-448. Abstract Article
Luzio, J. P., Pryor, P. R., and Bright, N. A. (2007). Lysosomes: fusion and function. Nat. Rev. Mol. Cell Biol. 8, 622-632. Abstract Article
Maiuri, M. C., Criollo, A., Tasdemir, E., Vicencio, J. M., Tajeddine, N., Hickman, J. A., Geneste, O., and Kroemer, G. (2007a). BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L). Autophagy 3, 374-376. Abstract
Maiuri, M. C., Le Toumelin, G., Criollo, A., Rain, J. C., Gautier, F., Juin, P., Tasdemir, E., Pierron, G., Troulinaki, K., Tavernarakis, N., et al. (2007b). Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 26, 2527-2539. Abstract Article
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007c). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741-752. Abstract Article
Majeski, A. E., and Dice, J. F. (2004). Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 36, 2435-2444. Abstract Article
Mammucari, C., Milan, G., Romanello, V., Masiero, E., Rudolf, R., Del Piccolo, P., Burden, S. J., Di Lisi, R., Sandri, C., Zhao, J., et al. (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 6, 458-471. Abstract Article
Mammucari, C., Schiaffino, S., and Sandri, M. (2008). Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 4, 524-526. Abstract
Massey, A., Kiffin, R., and Cuervo, A. M. (2004). Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 36, 2420-2434. Abstract Article
Matsuura, A., Tsukada, M., Wada, Y., and Ohsumi, Y. (1997). Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192, 245-250. Abstract Article
Meijer, A. J., and Codogno, P. (2004). Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 36, 2445-2462. Abstract Article
Meijer, A. J., and Codogno, P. (2006). Signalling and autophagy regulation in health, aging and disease. Mol. Aspects Med. 27, 411-425. Abstract Article
Meijer, A. J., and Codogno, P. (2007). AMP-activated protein kinase and autophagy. Autophagy 3, 238-240. Abstract
Meléndez, A., Hall, D.H. and Hansen, M.(2008). Monitoring the role of autophagy in C. elegans aging. Methods Enzymol. 451, 493-520. Abstract Article
Meléndez, A., Tallóczy, Z., Seaman, M., Eskelinen, E. L., Hall, D. H., and Levine, B. (2003). Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387-1391. Abstract Article
Mihaylova, V. T., Borland, C. Z., Manjarrez, L., Stern, M. J., and Sun, H. (1999). The PTEN tumor suppressor homolog in Caenorhabditis elegans regulates longevity and dauer formation in an insulin receptor-like signaling pathway. Proc. Natl. Acad. Sci. USA 96, 7427-7432. Abstract Article
Mills, K. R., Reginato, M., Debnath, J., Queenan, B., and Brugge, J. S. (2004). Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc. Natl. Acad. Sci. USA 101, 3438-3443. Abstract Article
Mizushima, N. (2004). Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 36, 2491-2502. Abstract Article
Mizushima, N., and Klionsky, D. J. (2007). Protein turnover via autophagy: implications for metabolism. Annu. Rev. Nutr. 27, 19-40. Abstract Article
Mizushima, N., Kuma, A., Kobayashi, Y., Yamamoto, A., Matsubae, M., Takao, T., Natsume, T., Ohsumi, Y., and Yoshimori, T. (2003). Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J. Cell Sci. 116, 1679-1688. Abstract Article
Mizushima, N., Noda, T., and Ohsumi, Y. (1999). Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 18, 3888-3896. Abstract Article
Mizushima, N., Noda, T., Yoshimori, T., Tanaka, Y., Ishii, T., George, M. D., Klionsky, D. J., Ohsumi, M., and Ohsumi, Y. (1998). A protein conjugation system essential for autophagy. Nature 395, 395-398. Abstract Article
Mizushima, N., Yamamoto, A., Hatano, M., Kobayashi, Y., Kabeya, Y., Suzuki, K., Tokuhisa, T., Ohsumi, Y., and Yoshimori, T. (2001). Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 152, 657-668. Abstract Article
Mizushima, N., Yamamoto, A., Matsui, M., Yoshimori, T., and Ohsumi, Y. (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 15, 1101-1111. Abstract Article
Mörck, C., and Pilon, M. (2006). C. elegans feeding defective mutants have shorter body lengths and increased autophagy. BMC Dev. Biol. 6, 39. Abstract
Morley, J. F., Brignull, H. R., Weyers, J. J., and Morimoto, R. I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 99, 10417-10422. Abstract Article
Morris, J. Z., Tissenbaum, H. A., and Ruvkun, G. (1996). A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382, 536-539. Abstract Article
Muro, I., Berry, D. L., Huh, J. R., Chen, C. H., Huang, H., Yoo, S. J., Guo, M., Baehrecke, E. H., and Hay, B. A. (2006). The Drosophila caspase Ice is important for many apoptotic cell deaths and for spermatid individualization, a nonapoptotic process. Development 133, 3305-3315. Abstract Article
Murphy, M. P., and Golde, T. E. (2006). Inclusion-body myositis and Alzheimer disease: two sides of the same coin, or different currencies altogether? Neurology 66, S65-68. Abstract Article
Neufeld, T. P. (2003). Body building: regulation of shape and size by PI3K/TOR signaling during development. Mech. Dev. 120, 1283-1296. Abstract Article
Neufeld, T. P. (2007). Contribution of Atg1-Dependent autophagy to TOR-mediated cell growth and survival. Autophagy 3, 477-479. Abstract
Nicot, A. S., Fares, H., Payrastre, B., Chisholm, A. D., Labouesse, M., and Laporte, J. (2006). The phosphoinositide kinase PIKfyve/Fab1p regulates terminal lysosome maturation in Caenorhabditis elegans. Mol. Biol. Cell 17, 3062-3074. Abstract
Nishino, I., Fu, J., Tanji, K., Yamada, T., Shimojo, S., Koori, T., Mora, M., Riggs, J. E., Oh, S. J., Koga, Y., et al. (2000). Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906-910. Abstract Article
Noda, T., Kim, J., Huang, W. P., Baba, M., Tokunaga, C., Ohsumi, Y., and Klionsky, D. J. (2000). Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J. Cell Biol. 148, 465-480. Abstract Article
Noda, T., and Ohsumi, Y. (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 273, 3963-3966. Abstract Article
Ogg, S., Paradis, S., Gottlieb, S., Patterson, G. I., Lee, L., Tissenbaum, H. A., and Ruvkun, G. (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994-999. Abstract Article
Ogg, S., and Ruvkun, G. (1998). The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol. Cell 2, 887-893. Abstract Article
Ogura, K., and Goshima, Y. (2006). The autophagy-related kinase UNC-51 and its binding partner UNC-14 regulate the subcellular localization of the Netrin receptor UNC-5 in Caenorhabditis elegans. Development 133, 3441-3450. Abstract Article
Ogura, K., Wicky, C., Magnenat, L., Tobler, H., Mori, I., Müller, F., and Ohshima, Y. (1994). Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev. 8, 2389-2400. Abstract Article
Oh, S. W., Mukhopadhyay, A., Svrzikapa, N., Jiang, F., Davis, R. J., and Tissenbaum, H. A. (2005). JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA 102, 4494-4499. Abstract Article
Panowski, S. H., Wolff, S., Aguilaniu, H., Durieux, J., and Dillin, A. (2007). PHA-4/Foxa mediates diet-restriction-induced longevity of C. elegans. Nature 447, 550-555. Abstract Article
Paradis, S., Ailion, M., Toker, A., Thomas, J. H., and Ruvkun, G. (1999). A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 13, 1438-1452. Abstract Article
Paradis, S., and Ruvkun, G. (1998). Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 12, 2488-2498. Abstract Article
Patterson, G. I., and Padgett, R. W. (2000). TGFβ-related pathways. Roles in Caenorhabditis elegans development. Trends Genet. 16, 27-33. Abstract Article
Pattingre, S., Bauvy, C., Carpentier, S., Levade, T., Levine, B., and Codogno, P. (2009). Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 284, 2719-2728. Abstract Article
Pattingre, S., Bauvy, C., and Codogno, P. (2003a). Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 278, 16667-16674. Abstract Article
Pattingre, S., De Vries, L., Bauvy, C., Chantret, I., Cluzeaud, F., Ogier-Denis, E., Vandewalle, A., and Codogno, P. (2003b). The G-protein regulator AGS3 controls an early event during macroautophagy in human intestinal HT-29 cells. J. Biol. Chem. 278, 20995-21002. Abstract Article
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., Packer, M., Schneider, M. D., and Levine, B. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927-939. Abstract Article
Petiot, A., Ogier-Denis, E., Blommaart, E. F., Meijer, A. J., and Codogno, P. (2000). Distinct classes of phosphatidylinositol 3'-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 275, 992-998. Abstract Article
Qu, X., Zou, Z., Sun, Q., Luby-Phelps, K., Cheng, P., Hogan, R. N., Gilpin, C., and Levine, B. (2007). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931-946. Abstract Article
Raizen, D. M., Cullison, K. M., Pack, A. I., and Sundaram, M. V. (2006). A novel gain-of-function mutant of the cyclic GMP-dependent protein kinase egl-4 affects multiple physiological processes in Caenorhabditis elegans. Genetics 173, 177-187. Abstract Article
Rea, S. L., Ventura, N., and Johnson, T. E. (2007). Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans. PLoS Biol. 5, e259. Abstract Article
Reggiori, F., and Klionsky, D. J. (2006). Atg9 sorting from mitochondria is impaired in early secretion and VFT-complex mutants in Saccharomyces cerevisiae. J. Cell Sci. 119, 2903-2911. Abstract Article
Reggiori, F., Tucker, K. A., Strømhaug, P. E., and Klionsky, D. J. (2004). The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev. Cell 6, 79-90. Abstract Article
Ren, P., Lim, C. S., Johnsen, R., Albert, P. S., Pilgrim, D., and Riddle, D. L. (1996). Control of C. elegans larval development by neuronal expression of a TGF-β homolog. Science 274, 1389-1391. Abstract Article
Riddle, D. L. and Albert, P. S. (1997). Genetic and environmental regulation of dauer larva development. In C. elegans II, D. L. Riddle, T. Blumenthal, B. J. Meyer, and J. R. Priess, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp. 739-768.
Roggo, L., Bernard, V., Kovács, A. L., Rose, A. M., Savoy, F., Zetka, M., Wymann, M. P., and Müller, F. (2002). Membrane transport in Caenorhabditis elegans: an essential role for VPS34 at the nuclear membrane. EMBO J. 21, 1673-1683. Abstract Article
Rouault, J. P., Kuwabara, P. E., Sinilnikova, O. M., Duret, L., Thierry-Mieg, D., and Billaud, M. (1999). Regulation of dauer larva development in Caenorhabditis elegans by daf-18, a homologue of the tumour suppressor PTEN. Curr. Biol. 9, 329-332. Abstract Article
Rowland, A. M., Richmond, J. E., Olsen, J. G., Hall, D. H., and Bamber, B. A. (2006). Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J. Neurosci. 26, 1711-1720. Abstract Article
Rusten, T. E., Lindmo, K., Juhász, G., Sass, M., Seglen, P. O., Brech, A., and Stenmark, H. (2004). Programmed autophagy in the Drosophila fat body is induced by ecdysone through regulation of the PI3K pathway. Dev. Cell 7, 179-192. Abstract Article
Samara, C., Syntichaki, P., and Tavernarakis, N. (2008). Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 15, 105-112. Abstract Article
Savage, C., Das, P., Finelli, A. L., Townsend, S. R., Sun, C. Y., Baird, S. E., and Padgett, R. W. (1996). Caenorhabditis elegans genes sma-2, sma-3, and sma-4 define a conserved family of transforming growth factor beta pathway components. Proc. Natl. Acad. Sci. USA 93, 790-794. Abstract Article
Scott, R. C., Juhász, G., and Neufeld, T. P. (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17, 1-11. Abstract Article
Scott, R. C., Schuldiner, O., and Neufeld, T. P. (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7, 167-178. Abstract Article
Seaman, M. N., Marcusson, E. G., Cereghino, J. L., and Emr, S. D. (1997). Endosome to Golgi retrieval of the vacuolar protein sorting receptor, Vps10p, requires the function of the VPS29, VPS30, and VPS35 gene products. J. Cell Biol. 137, 79-92. Abstract Article
Seglen, P. O., and Gordon, P. B. (1982). 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 79, 1889-1892. Abstract Article
Shacka, J. J., and Roth, K. A. (2005). Cathepsin deficiency as a model for neuronal ceroid lipofuscinoses. Am. J. Pathol. 167, 1473-1476. Abstract
Shintani, T., Mizushima, N., Ogawa, Y., Matsuura, A., Noda, T., and Ohsumi, Y. (1999). Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 18, 5234-5241. Abstract Article
Shintani, T., Suzuki, K., Kamada, Y., Noda, T., and Ohsumi, Y. (2001). Apg2p functions in autophagosome formation on the perivacuolar structure. J. Biol. Chem. 276, 30452-30460. Abstract Article
Suzuki, K., Kirisako, T., Kamada, Y., Mizushima, N., Noda, T., and Ohsumi, Y. (2001). The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971-5981. Abstract Article
Suzuki, K., Kubota, Y., Sekito, T., and Ohsumi, Y. (2007). Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209-218. Abstract Article
Suzuki, K., and Ohsumi, Y. (2007). Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 581, 2156-2161. Abstract Article
Syntichaki, P., Xu, K., Driscoll, M., and Tavernarakis, N. (2002). Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419, 939-944. Abstract Article
Takács-Vellai, K., Vellai, T., Puoti, A., Passannante, M., Wicky, C., Streit, A., Kovács, A. L., and Müller, F. (2005). Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr. Biol. 15, 1513-1517. Abstract Article
Takahashi, Y., Coppola, D., Matsushita, N., Cualing, H. D., Sun, M., Sato, Y., Liang, C., Jung, J. U., Cheng, J. Q., Mul, J. J., et al. (2007). Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 9, 1142-1151. Abstract Article
Takeshige, K., Baba, M., Tsuboi, S., Noda, T., and Ohsumi, Y. (1992). Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301-311. Abstract Article
Tallóczy, Z., Jiang, W., Virgin, H. W. 4th, Leib, D. A., Scheuner, D., Kaufman, R. J., Eskelinen, E. L., and Levine, B. (2002). Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc. Natl. Acad. Sci. USA 99, 190-195. Abstract Article
Tanida, I., Tanida-Miyake, E., Komatsu, M., Ueno, T., and Kominami, E. (2002). Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J. Biol. Chem. 277, 13739-13744. Abstract Article
Teter, S. A., Eggerton, K. P., Scott, S. V., Kim, J., Fischer, A. M., and Klionsky, D. J. (2001). Degradation of lipid vesicles in the yeast vacuole requires function of Cvt17, a putative lipase. J. Biol. Chem. 276, 2083-2087. Abstract
Thomas, J. H., Birnby, D. A., and Vowels, J. J. (1993). Evidence for parallel processing of sensory information controlling dauer formation in Caenorhabditis elegans. Genetics 134, 1105-1117. Abstract
Tooze, J., Hollinshead, M., Ludwig, T., Howell, K., Hoflack, B., and Kern, H. (1990). In exocrine pancreas, the basolateral endocytic pathway converges with the autophagic pathway immediately after the early endosome. J. Cell Biol. 111, 329-345. Abstract Article
Tóth, M. L., Sigmond, T., Borsos, E., Barna, J., Erdélyi, P., Takács-Vellai, K., Orosz, L., Kovács, A. L., Csikos, G., Sass, M., and Vellai, T. (2008). Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 4, 330-338. Abstract
Tóth, M. L., Simon, P., Kovács, A. L., and Vellai, T. (2007). Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J. Cell Sci. 120, 1134-1141. Abstract Article
Treinin, M., and Chalfie, M. (1995). A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 14, 871-877. Abstract Article
Tucker, K. A., Reggiori, F., Dunn, W. A., Jr., and Klionsky, D. J. (2003). Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J. Biol. Chem. 278, 48445-48452. Abstract Article
Vellai, T., Takács-Vellai, K., Zhang, Y., Kovács, A. L., Orosz, L., and Müller, F. (2003). Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426, 620. Abstract
Voisine, C., Varma, H., Walker, N., Bates, E. A., Stockwell, B. R., and Hart, A. C. (2007). Identification of potential therapeutic drugs for Huntington's disease using Caenorhabditis elegans. PLoS ONE 2, e504. Abstract Article
Wang, C. W., Kim, J., Huang, W. P., Abeliovich, H., Strømhaug, P. E., Dunn, W. A., Jr., and Klionsky, D. J. (2001). Apg2 is a novel protein required for the cytoplasm to vacuole targeting, autophagy, and pexophagy pathways. J. Biol. Chem. 276, 30442-30451. Abstract Article
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678-688. Abstract Article
White, J. G., Southgate E., Thomson, J. N., Brenner S. (1986). The structure of the nervous system of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 314, 1-340.
Xie, Z., and Klionsky, D. J. (2007). Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 9, 1102-1109. Abstract Article
Yen, W. L., and Klionsky, D. J. (2007). Atg27 is a second transmembrane cycling protein. Autophagy 3, 254-256. Abstract
Yen, W. L., Legakis, J. E., Nair, U., and Klionsky, D. J. (2007). Atg27 is required for autophagy-dependent cycling of Atg9. Mol. Biol. Cell 18, 581-593. Abstract Article
Yorimitsu, T., and Klionsky, D. J. (2005). Autophagy: molecular machinery for self-eating. Cell Death Differ. 12 Suppl 2, 1542-1552. Abstract
*Edited by James M. Kramer and Donald C. Moerman. Last revised February 22, 2009. Published August 24, 2009. This chapter should be cited as: Meléndez, A. and Levine, B. Autophagy in C. elegans (August 24, 2009), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.147.1, http://www.wormbook.org.
Copyright: © 2009 Alicia Meléndez and Beth Levine. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail : alicia.melendez@qc.cuny.edu or beth.levine@utsouthwestern.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.