Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Acetylcholine is the major excitatory neurotransmitter at nematode neuromuscular junctions, and more than a third of the cells in the C. elegans nervous system release acetylcholine. Through a combination of forward genetics, drug-resistance selections, and genomic analysis, mutants have been identified for all of the steps specifically required for cholinergic function. These include two enzymes, two transporters, and a bewildering assortment of receptors. Cholinergic transmission is involved, directly or indirectly, in many C. elegans behaviors, including locomotion, egg laying, feeding, and male mating.
Acetylcholine (ACh) was the first substance proven to be a neurotransmitter (Loewi, 1921). It was identified in Ascaris and other nematodes in 1955 by Helen Mellanby (Mellanby, 1955), and was subsequently shown to be an excitatory transmitter at nematode neuromuscular junctions (del Castillo et al., 1963; del Castillo et al., 1967).
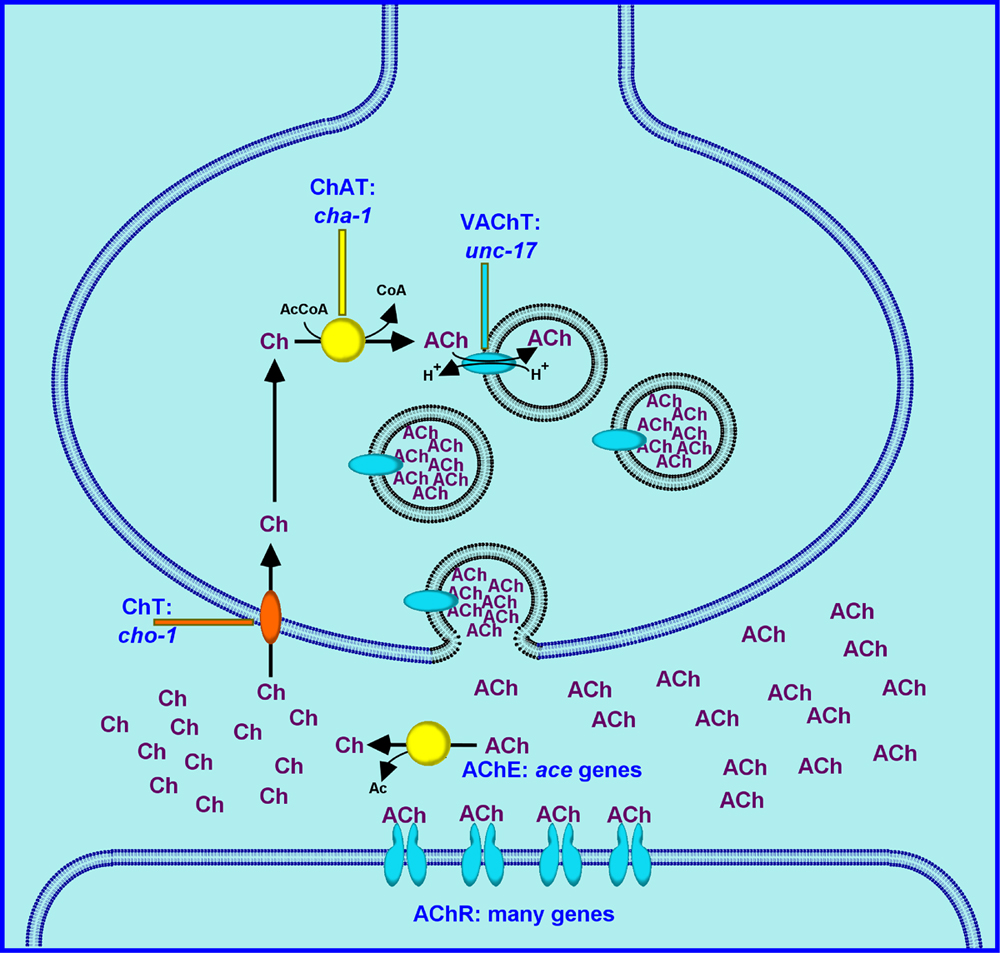
As shown in Figure 1, ACh is synthesized by choline acetyltransferase (ChAT), and is loaded into synaptic vesicles by the vesicular acetylcholine transporter (VAChT). The synaptic vesicle lumen is acidified by the action of an ATP-dependent proton pump located in the synaptic vesicle membrane. The pH gradient between the vesicle lumen and the cytoplasm provides the driving force for ACh transport; the VAChT essentially “exchanges” ACh for protons. The docking and priming of synaptic vesicles, and their calcium-stimulated fusion with the cell membrane are all general processes that are independent of the neurotransmitter contained in the vesicles (and are described elsewhere in this volume). Following synaptic vesicle fusion and transmitter release, the ACh diffuses within the synaptic cleft and activates acetylcholine receptors (AChRs), usually located on post-synaptic cells. For most other neurotransmitters (e.g., GABA, dopamine, serotonin), the action of the transmitter is terminated by transporter- mediated removal of the transmitter from the synaptic cleft. The action of acetylcholine, however, is terminated by direct enzymatic hydrolysis of the neurotransmitter in the synaptic cleft by acetylcholinesterase (AChE). The resulting choline is then transported back into the presynaptic neuron by a high affinity choline transporter (HAChT, or ChT); this choline is then available for the synthesis of additional ACh.
In subsequent sections, each of these steps will be described, along with properties and subcellular localization of the proteins, their expression pattern, and mutant phenotypes. I then discuss ACh-mediated behaviors, and some additional aspects of cholinergic biology.
Sydney Brenner first reported the isolation of drug-resistant mutants of C. elegans; the drugs he tested were the cholinesterase inhibitor lannate and the acetylcholine receptor agonist tetramisole (Brenner, 1974). Since then, the use of cholinergic drugs for mutant isolation has been extremely useful. Many of the mutations identified by drug resistance turned out to be alleles of genes previously identified as Uncs (e.g., unc-17, unc-29), yet a significant number of the drug resistance mutations were in previously unidentified loci (e.g., snt-1, ric-8, lev-1).
Mutations in a large number of genes can confer resistance, and for many of these genes, null alleles are viable; thus mutant screens and selections have been quite productive (Lewis et al., 1980b; Nguyen et al., 1995; Miller et al., 1996). Many cholinergic drugs (some of which are mentioned in subsequent sections) have been used in the analysis of specific mutants and behaviors; however, the two that have had the most significant impact on C. elegans neurobiology are aldicarb and levamisole.
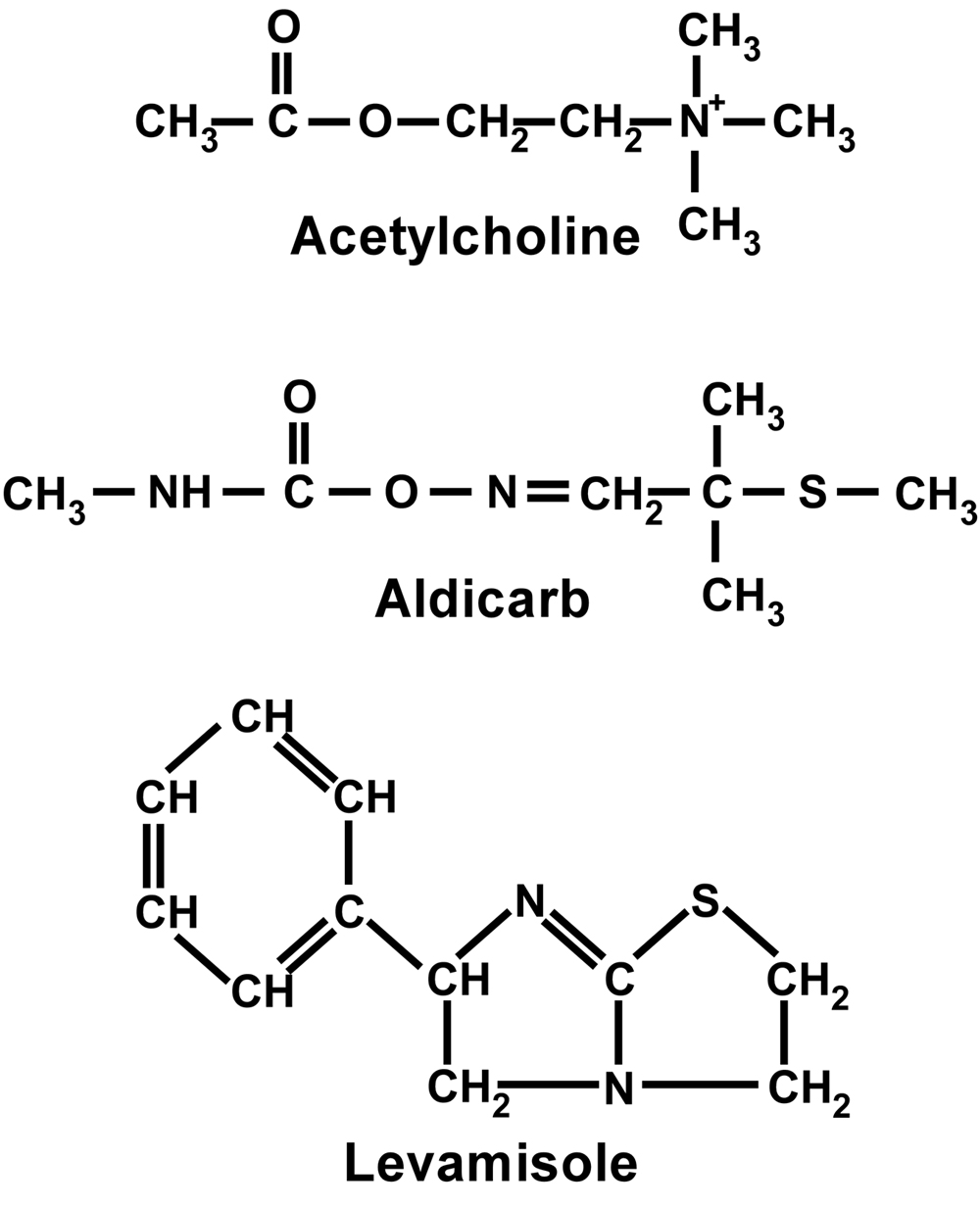
Although lannate was initially used for such studies (Brenner, 1974), and trichlorfon has also been used (Hosono et al., 1989), the carbamate acetylcholinesterase inhibitor aldicarb (see Figure 2) has become the reagent of choice for evaluation of the “Ric” phenotype (for Resistance to Inhibitors of Cholinesterase). There are more than 50 genes known which can mutate to confer a Ric phenotype. The direct consequence of inhibiting AChE is a buildup of synaptic ACh, and since ACh is the excitatory transmitter at neuromuscular junctions, the excess ACh causes a hypercontracted paralysis. In principle, there should be two major gene classes which can mutate to confer aldicarb resistance. A mutation which disrupts ACh reception or any portion of the postsynaptic response to ACh would be expected to confer resistance (the muscle wouldn't “know” or “care” that there was excess ACh on its doorstep). In addition, however, any mutation which disrupted ACh synthesis, vesicular loading or release would lead to less ACh being released by the motor neurons, so there wouldn't be as much ACh to build up in the first place. A third possibility, mutation of an acetylcholinesterase subunit, would presumably require a very specific gain-of-function mutation, and is expected to be extremely rare.
Operationally, there are two strategies to determine/assess resistance to aldicarb. One can measure behavioral impairment following “acute” exposure to aldicarb, for example, the percent of animals totally paralyzed after 4 hrs as a function of aldicarb concentration (Nonet et al., 1993), or the percent of animals totally paralyzed as a function of time at, e.g., 2 mM aldicarb (Schade et al., 2005). An alternative is to measure growth and development in the presence of aldicarb, i.e., “chronic” exposure (Nguyen et al., 1995; Miller et al., 1999). Most mutageneses have employed a selection scheme based on growth and reproduction in the presence of aldicarb.
Although a few of the Ric mutations clearly affect cholinergic targets (e.g., cha-1, unc-17, ric-3, described in subsequent sections), most of them are in genes involved in the release of all neurotransmitters. Thus, for example, the first mutations in snt-1 (synaptotagmin), snb-1 (synaptobrevin), and ric-8 (synembryn) were isolated in large-scale selections for aldicarb resistance (Nonet et al., 1993; Nonet et al., 1998; Miller et al., 2000). As a result, aldicarb resistance in C. elegans is often equated with decreased neurotransmitter release, an unfortunate oversimplification. Nevertheless, a number of successful forward genetic screens have identified many important presynaptic components (Nguyen et al., 1995; Miller et al., 1996; Yook et al., 2001), and a recent genome-scale reverse genetic approach using aldicarb to identify genes affecting synaptic function has been described (Sieburth et al., 2005).
In contrast to the Ric phenotype, mutants have been described with the opposite “Hic” phenotype (Hypersensitivity to Inhibitors of Cholinesterase); and while many mutants with reductions in transmitter release are associated with aldicarb resistance, a number of mutants with apparent elevated release of acetylcholine are characterized by hypersensitivity to aldicarb (Lackner et al., 1999; Miller et al., 1999; Nurrish et al., 1999; Miller and Rand, 2000; Reynolds et al., 2005; Schade et al., 2005).
A particularly fruitful use of aldicarb mutants has been in the elaboration of a G-protein-coupled regulatory network that regulates ACh release from the ventral cord motor neurons (see Heterotrimeric G proteins in C. elegans). A core pathway in this network includes RIC-8, Gαq (EGL-30) and phospholipase Cβ (EGL-8) and leads to production of DAG, which stimulates transmitter release; loss-of-function mutations in these components lead to decreased locomotion, decreased egg laying, and aldicarb resistance (Miller et al., 1999; Lackner et al., 1999; Miller et al., 2000; Reynolds et al., 2005). Conversely, gain-of-function mutations in this core pathway lead to the opposite phenotypes: hyperactive locomotion and egg laying, and hypersensitivity to aldicarb. Negative regulators of this pathway include Gα0 (GOA-1) and DAG kinase (DGK-1), which reduce DAG levels and reduce transmitter release; loss-of-function mutations in such negative regulators lead to hyperactivity and aldicarb hypersensitivity (Nurrish et al., 1999). Many of the mutants that defined this signaling network were either isolated on the basis of altered response to aldicarb, or their response to aldicarb provided critical data for the interpretation of their function(s).
Levamisole (see Figure 2) is a potent cholinergic agonist, and is often used as a pesticide. It leads to a hypercontracted paralysis of wild-type nematodes, usually followed by relaxation and death (Lewis et al., 1980b). Levamisole has been shown to bind specifically to one of the two receptor types present in body-wall muscle, and has therefore been an extremely valuable tool in the molecular and physiological analysis of C. elegans AChR subunits (see Behavior section of WormMethods) . Operationally, the sensitivity or resistance of a given strain may be determined by monitoring either the time-course of paralysis or the extent of body-shortening due to muscle hypercontraction. The majority of levamisole-resistance genes were identified in several large-scale mutageneses by Jim Lewis (Lewis et al., 1980b; Lewis et al., 1980a). Mutations in several genes conferred strong resistance to levamisole, and also a mild uncoordinated phenotype: the AChR subunit-encoding genes unc-29, unc-38, and unc-63, and the accessory protein encoding genes unc-50 and unc-74 (Lewis et al., 1980b). There were also mutations conferring mild resistance to levamisole and normal locomotion; these identified the receptor subunit-encoding genes lev-1 and lev-8, and the accessory protein encoding gene lev-10. These genes are all described below in Sections 6 and 8.
Choline acetyltransferase catalyzes the acetylation of choline by acetyl-Coenzyme A (see Figure 1). The C. elegans enzyme has been partially purified and characterized (Rand and Russell, 1985); it is similar to the vertebrate and Drosophila ChAT proteins. Immunolocalization studies indicate that most of the ChAT protein is synaptic, but some immunoreactivity appears to be generally cytoplasmic (J. Duerr and J. Rand, unpublished). The synaptic protein is associated with a vesicular compartment, most likely synaptic vesicles, because it was mislocalized in unc-104 mutants. Both the synaptic and the cytoplasmic staining were decreased in particular cha-1 mutant strains and increased in transgenic lines which overexpress ChAT (J. Duerr and J. Rand, unpublished).
The vesicular acetylcholine transporter was first cloned and characterized in C. elegans (Alfonso et al., 1993), where it was shown to be the product of the unc-17 gene. Structurally, the UNC-17 (VAChT) protein has 12 transmembrane domains, with cytoplasmic (i.e., extravesicular) N- and C-termini, and is closely related to the vesicular monoamine transporters (VMATs). Immunoreactivity with anti-UNC-17 antibody preparations in wild-type animals is punctate and colocalizes with the synaptic vesicle protein synaptotagmin, suggesting that UNC-17 is associated with synaptic vesicles (Alfonso et al., 1993). This punctate staining is mislocalized in unc-104 mutants, providing additional support to the conclusion that UNC-17 is an integral membrane protein of synaptic vesicles.
Although there are several formal criteria used by neurobiologists to determine if a neuron is cholinergic, the unofficial criterion is expression of ChAT. Operationally, this means either immunolocalization of ChAT or expression of a cholinergic reporter (discussed below). By such criteria, approximately 120 neurons in the C. elegans hermaphrodite are cholinergic. The majority of these cells are motor neurons. These include the AS, DA, DB, VA, VB, and VC motor neuron classes of the ventral nerve cord, the sublateral motor neuron classes SAA, SAB, SIA, SIB, SMB, and SMD (Rand and Nonet, 1997), and the HSN cells (Duerr et al., 2001).
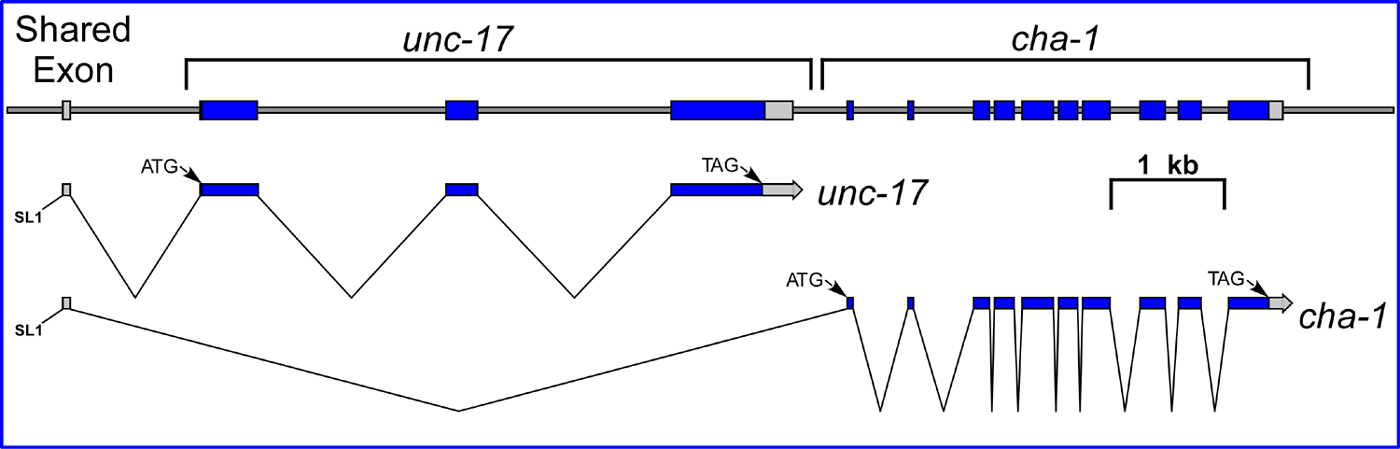
Genetic and molecular studies showed that cha-1 and unc-17 were part of a complex gene locus and transcription unit, with the unc-17 gene nested in the long first intron of cha-1 (see Figure 3; Rand, 1989; Alfonso et al., 1994a). Thus, the sequential steps of acetylcholine synthesis and vesicle-loading are encoded by different genes within a single, complex transcription unit. Subsequent studies demonstrated that a similar nested structure of these genes is present in mammals and insects (Erickson et al., 1994; Kitamoto et al., 1998). This is now commonly called the cholinergic gene locus, or CGL, and is present in all metazoans thus far examined (Eiden, 1998). It is noteworthy that the genes encoding enzymes and transporters for other neurotransmitters are not organized in a comparable way.
As described below, the unc-17 promoter regulates the expression of both genes, and may therefore be considered as a “cholinergic” promoter.
unc-17 mutants were first described by Brenner (Brenner, 1974), and cha-1 mutants were described 10 years later (Rand and Russell, 1984). More than a dozen cha-1 alleles and more than 20 unc-17 alleles have now been described (Brenner, 1974; Rand and Russell, 1984; Hosono et al., 1985; Rand, 1989; Alfonso et al., 1993; Alfonso et al., 1994a; Alfonso et al., 1994b; Zhao and Nonet, 2000; Zhu et al., 2001). Hypomorphic cha-1 mutants (including animals homozygous for the reference p1152 allele) are coily, uncoordinated, and jerky in reverse; they are also small, slow-growing, and resistant to aldicarb (Rand and Russell, 1984). Null alleles of cha-1 are lethal - the animals hatch, but they are essentially immobile, and do not grow or feed (Rand, 1989; Alfonso et al., 1993). In general, the phenotypes of hypomorphic unc-17 mutants (including animals homozygous for the e245 reference allele) are similar to those of cha-1: animals are small, slow-growing, coiling Uncs, jerky in reverse, and resistant to cholinesterase inhibitors (Brenner, 1974; Rand and Russell, 1984). Null unc-17 mutants are lethal, with a phenotype quite similar to cha-1 null mutants (Alfonso et al., 1993). The lethality of unc-17 null mutants and the similarity of their phenotype to cha-1 nulls argue that vesicular loading is an obligatory step in ACh release.
The molecular analysis of the region has helped to explain some genetic anomalies. Even though in general, cha-1 mutations and unc-17 mutations behaved as two discrete complementation groups, there were a few mutations that failed to complement both cha-1 and unc-17 alleles (Rand and Russell, 1984). These fell into two classes, termed α and β. Fine-structure genetic mapping indicated that the single α allele mapped between cha-1 and unc-17, but the two β alleles mapped well to the right of both loci (Rand, 1989). Subsequent molecular analysis revealed that the α allele corresponds to a small deletion between unc-17 and cha-1 (Alfonso et al., 1994a), and the β mutations are in the promoter region upstream of the unc-17 coding sequence (D. Frisby and J. Rand, unpublished). The observed noncomplementation between the β mutations and cha-1 alleles confirms that the promoter region upstream of unc-17 drives both unc-17 and cha-1 transcription (see Figure 3).
Like the vesicular acetylcholine transporter, the plasma membrane choline transporter was first cloned and characterized in C. elegans (Okuda et al., 2000). The ChT protein (CHO-1) is a member of a class of sodium-dependent transporters with substrates such as glucose (SGLT1) and inositol (SMIT1). It is best modeled as a 13-transmembrane domain protein, with a short N-terminus oriented extracellularly, and an extended, C-terminus oriented into the cytoplasm (Apparsundaram et al., 2000; Wang et al., 2001). Assays performed in embryonic cell culture showed that the C. elegans CHO-1 transporter was sodium- and chloride-dependent, with an apparent Km for choline of 0.66 μM, comparable to mammalian transporters (Matthies et al., 2006).
Reporter studies indicate that cho-1 is expressed in most or all cholinergic neurons (Matthies et al., 2006). Within these neurons, a CHO-1-GFP fusion protein was localized to synaptic regions and was associated with a vesicular compartment, because its localization was dependent on UNC-104 (Matthies et al., 2006). This is consistent with results from mammalian systems demonstrating that the choline transporter is localized to apparent synaptic vesicles (Ferguson et al., 2003).
The reference allele, tm373, is a precise 1695 bp deletion which eliminates more than half of the cho-1 coding sequence; it is almost certainly a null allele. Animals homozygous for tm373 are viable, and their growth and development appear to be normal. However, although cho-1 mutants have little difficulty crawling on agar, they swim somewhat more slowly that wild-type animals, and become paralyzed (“fatigued”) more quickly than wild-type animals during prolonged swimming in liquid (Matthies et al., 2006). It is tempting to speculate that there is enough endogenous choline and/or low-affinity choline uptake activity (see below) in the cho-1 mutant nerve terminals to support some ACh synthesis even without any high-affinity reuptake activity, and that this level of ACh synthesis is sufficient for locomotion in a calm environment. However, this level of ACh is not adequate to support the increased demands of vigorous swimming, and the cho-1 motor neurons appear to become depleted of transmitter. This provides genetic-based evidence for a functional coupling of choline transport and ACh synthesis, and supports previous models based on other methods (Jope and Jenden, 1980; Collier, 1988; Bussiere et al., 2001).
If choline is indeed rate-limiting for acetylcholine synthesis under some circumstances, what determines the availability of choline? In C. elegans, choline is synthesized in the form of phosphocholine through progressive N-methylation of phosphoethanolamine by the PMT-1 and PMT-2 methyltransferases (Palavalli et al., 2006). Choline may also be produced through cleavage of phosphatidylcholine by phospholipase D (PLD-1). Choline is thus a product of lipid metabolism and lipid turnover. The activities of these enzymes have not been analyzed in cholinergic neurons.
SNF-6 is a post-synaptic choline/acetylcholine transporter (Kim et al., 2004). It is a member of the family of plasma membrane transporters which includes the serotonin transporter (MOD-5), the dopamine transporter (DAT-1), and the GABA transporter (SNF-11). The only two substrates identified for SNF-6 were choline and acetylcholine; both substrates had comparable Km values, but the Vmax for choline was about seven times the Vmax for acetylcholine (Kim et al., 2004). This appears to be a nematode-specific transport activity. The SNF-6 protein is expressed in muscles, and is preferentially localized to neuromuscular junctions. snf-6 mutants are somewhat uncoordinated and mildly hypersensitive to aldicarb. This transport activity may provide a mechanism for rapid clearance of synaptic ACh and/or choline during periods of high transmitter release.
Most species (and apparently C. elegans as well) also express several “low-affinity” choline transport activities with broad tissue distributions. The CTL proteins in mammals (corresponding to the chtl-1 gene product) are sodium-independent transporters of choline and other organic cations (O'Regan et al., 2000). There is also a family of at least five organic cation transport (OCT) proteins in mammals with broad, overlapping substrate specificities (including choline; Friedrich et al., 2001). C. elegans members of this family include OCT-1 (Wu et al., 1999), OCT-2 (listed in WormBase with no data), plus 3 or 4 others in WormPep which are mostly uncharacterized. The involvement, if any, of these transport activities in cholinergic function is unknown.
Biochemical analysis of C. elegans homogenates led to a complicated assortment of AChE forms with different molecular weights, subunit composition, and hydrophobicity; however, three classes of AChE activity (A, B, and C) could be distinguished on the basis of substrate affinity, inhibitor specificities, and detergent sensitivities (Johnson and Russell, 1983; Kolson and Russell, 1985). Vertebrates express two families of cholinesterase activity, the “true” acetylcholinesterases and the “pseudo” or butyrylcholinesterases. However, the three C. elegans AChE activity classes do not correspond in any clear way to the vertebrate enzyme classes (Johnson and Russell, 1983). The active site-containing subunits associated with the A, B, and C classes have been shown to be the gene products of the ace-1, ace-2, and ace-3 genes, respectively (Johnson et al., 1981; Culotti et al., 1981; Kolson and Russell, 1985; Combes et al., 2000). A fourth gene, ace-4, has been described, but the protein it encodes appears not to be enzymatically active (see below).
The three AChE classes were subsequently shown to include a number of forms with different association states. Class A (ACE-1) subunits self-associate, forming amphiphilic tetramers; these tetramers may also bind to a hydrophobic noncatalytic subunit which is poorly characterized (Combes et al., 2000). Class B (ACE-2) and Class C (ACE-3) subunits form glycolipid-anchored homodimers (Combes et al., 2000). The Class C AChE activity represents only a few percent of the total C. elegans AChE activity (Kolson and Russell, 1985), yet it is noteworthy in several ways. Its apparent Km of 16-18 nM is approximately 3 to 4 orders of magnitude lower than Km values for Classes A and B, as well as vertebrate AChEs (Johnson and Russell, 1983; Kolson and Russell, 1985). The Class C activity is also quite resistant to cholinesterase inhibitors (Kolson and Russell, 1985). Class C-like AChE activity with these properties has been identified in other nematode species, but is not present in vertebrates (Kolson and Russell, 1985; Selkirk et al., 2005).
The three classes of AChE have quite different tissue and cellular expression patterns. Mosaic analysis was initially used to determine that ace-1 was expressed in muscles, and not (or very little) in neurons (Herman and Kari, 1985). Subsequent studies with reporter genes demonstrated expression in all body muscle cells, as well as the anal sphincter muscle, the four vm1 vulval muscles, the three pm5 pharyngeal muscles, and six neurons (2 OLL and 4 CEP; Culetto et al., 1999). Histochemical methods showed that the ACE-1 enzymatic activity was localized in or near the nerve ring and nerve cords and was presumably postsynaptic at neuromuscular junctions (Culotti et al., 1981).
Reporter analysis using an ace-2 promoter indicated expression in a number of neurons in head (including the 12 IL cells and others), neurons in the tail (PVC, PVQ, and PDA), the pm5 pharyngeal muscles, and hypodermal cells near the tip of the tail (hyp8, 9, 10, 11; Combes et al., 2003). Histochemical methods showed that the ACE-2 enzymatic activity was localized near the nerve ring and nerve cords, and also near the pharyngeointestinal valve, although the cells contributing to this pattern were not identified (Culotti et al., 1981; Combes et al., 2003).
Expression of an ace-3 reporter was observed in a few neurons in the head, a few neurons in the tail (probably PQR and PDA), the two lateral CAN cells, as well as in the pm3, pm4, pm5, and pm7 pharyngeal muscles. A dorsal row of body-wall muscle cells (the 2 dorso-medial hemiquadrants) was intensely labeled in larval stages but no longer detected in adults (Combes et al., 2003). Immunolocalization of Class C revealed staining in the nerve ring and CAN cells (there was also apparent artifactual staining of the pharyngeointestinal valve; Stern, 1986). The CAN staining in the cell processes was of a comparable intensity to the cell body, consistent with the enzyme activity being uniformly distributed in or along the plasma membrane.
It is noteworthy that although the three activities are expressed in different cell types (with the exception that all three classes are expressed in the pm5 pharyngeal muscles), all three are localized in or near the nerve ring and Classes A and B are localized in or near the ventral and dorsal nerve cords.
The three ace genes were mapped by genetic methods and are unlinked (Johnson et al., 1981; Culotti et al., 1981; Johnson et al., 1988). Subsequently, by comparing the acetylcholinesterase homologs in the genome to the known map positions of the three ace loci, it was possible to clone each of the ace genes, analyze their transcripts and expression patterns, and characterize the available mutations (Arpagaus et al., 1994; Grauso et al., 1998; Combes et al., 2000; Combes et al., 2003). Surprisingly, in the region predicted to encode ace-3 there were 2 AChE-encoding genes arranged in a two-gene operon (Grauso et al., 1998; Combes et al., 2003). Sequencing of an ace-3 mutation revealed that it was in the downstream gene; although the upstream gene (now known as ace-4) is transcribed, the encoded protein appears to be catalytically inactive (Combes et al., 2000).
Mutations have been described for ace-1, ace-2, and ace-3 (Johnson et al., 1981; Culotti et al., 1981; Johnson et al., 1988). Each of the single mutants is essentially wild-type, although ace-2 animals are hypersensitive to aldicarb. The ace-2; ace-3 and ace-3; ace-1 double mutants are also essentially wild-type (although ace-2; ace-3 animals are hypersensitive to aldicarb); however, ace-2; ace-1 animals are quite uncoordinated (Culotti et al., 1981). The ace-2; ace-3; ace-1 triple mutant is lethal - its embryonic development is apparently normal, but many of the animals do not hatch, and those that do hatch are paralyzed and developmentally arrested (Johnson et al., 1988). A potential caveat to this phenotypic analysis is that the ace-3 allele that was used was dc2, which was subsequently shown to be associated with a deletion disrupting both the ace-3 and ace-4 genes (Combes et al., 2000); thus the lethal phenotype is in reality associated with the quadruple ace-2; ace-3; ace-4; ace-1 genotype. However, since the ace-4 gene does not appear to produce a functional protein, it is likely the ace-2; ace-3; ace-1 triple mutant would have the reported lethal phenotype.
There does not appear to be any coordinated or compensatory regulation of the ace genes. In each of the three single ace mutants and each of the three double ace mutants, the remaining cholinesterase activities are present at their wild-type levels, and are not elevated (Johnson et al., 1988).
The “standard” vertebrate nicotinic type of AChR is a pentameric ligand-gated anion channel. The ACh-binding α subunits may associate into homomeric structures, or they may associate with closely related non-α subunits.
The initial group of C. elegans receptor subunits was identified by mutations conferring resistance to levamisole (Lewis et al., 1980a; Fleming et al., 1997), but the majority of receptor subunits have now been identified by genomic sequence analysis (Mongan et al., 1998; Jones and Sattelle, 2004). A review has been published recently (Jones and Sattelle, 2004), and Table 1 summarizes the current status of this field.
Table 1. C. elegans acetylcholine receptor subunits.
| AChR subunit | Groupa | Type | Cellular expressionb | Mutant phenotype | Comments | Citations |
|---|---|---|---|---|---|---|
| ACR-7 | ACR-16 | α | Mongan et al., 1998 | |||
| ACR-9 | ACR-16 | Non-α | Mongan et al., 1998 | |||
| ACR-10 | ACR-16 | α | Mongan et al., 1998 | |||
| ACR-11 | ACR-16 | α | Mongan et al., 1998 | |||
| ACR-14 | ACR-16 | Non-α | DA, DB, VB, AS, DD, HSN, VC4/5, AIY, head neurons, muscle, intestine | Mongan et al., 1998; Fox et al., 2005 | ||
| ACR-15 | ACR-16 | α | Mongan et al., 1998 | |||
| ACR-16 | ACR-16 | α | Body muscles, some neurons | NonUnc; but synthetic severe Unc with unc-29 or unc-63 | Nicotine- sensitive receptor subunit | Ballivet et al., 1996; Francis et al., 2005; Touroutine et al., 2005 |
| ACR-19 | ACR-16 | α | Mongan et al., 2002 | |||
| ACR-21 | ACR-16 | α | Mongan et al., 2002 | |||
| EAT-2 | ACR-16 | Non-α | Pharyngeal muscles | Slow pumping | McKay et al., 2004 | |
| ACR-6 | UNC-38 | α | Mongan et al., 1998 | |||
| UNC-38 | UNC-38 | α | Body and vulval muscles, many neurons | Unc, Lev | Levamisole- sensitive receptor subunit | Fleming et al., 1997; Gottschalk et al., 2005 |
| UNC-63 | UNC-38 | α | Body and vulval muscles, many neurons | Unc, Lev; synthetic severe Unc with acr-16 | Levamisole- sensitive receptor subunit | Culetto et al., 2004; Touroutine et al., 2005; Ruaud and Bessereau, 2006 |
| ACR-2 | UNC-29 | Non-α | VA, DA, VB, DB, IL1, RMD, and PVQ | Possible operon with acr-3 | Squire et al., 1995; Hallam et al., 2000; Y. Jin, cited in Nurrish et al., 1999 | |
| ACR-3 | UNC-29 | Non-α | Possible operon with acr-2 | Baylis et al., 1997 | ||
| LEV-1 | UNC-29 | Non-α | Body muscles, some neurons in ventral cord | Unc, Lev | Levamisole- sensitive receptor subunit | Fleming et al., 1997; Culetto et al., 2004 |
| UNC-29 | UNC-29 | Non-α | Body muscles, some neurons | Unc, Lev; synthetic severe Unc with acr-16 | Levamisole- sensitive receptor subunit | Fleming et al., 1997; Francis et al., 2005 |
| ACR-8 | ACR-8 | α | Body muscles, a few neurons in head and tail | Mongan et al., 1998; Touroutine et al., 2005; Gottschalk et al., 2005 | ||
| ACR-12 | ACR-8 | α | Exclusively in neurons, including ventral cord neurons | Mongan et al., 1998; Gottschalk et al., 2005 | ||
| LEV-8 | ACR-8 | α | Body muscles, uv1 and uv2 uterine muscles, PVT, ALA, many ventral cord cells including all DD cells, many head neurons, IL and OL socket cells | Lev | =ACR-13; levamisole- sensitive receptor subunit | Mongan et al., 1998; Towers et al., 2005 |
| ACR-5 | DEG-3 | α | DB, VB motor neurons; other neurons in head and tail | Mongan et al., 1998; Winnier et al., 1999 | ||
| ACR-17 | DEG-3 | α | Mongan et al., 2002 | |||
| ACR-18 | DEG-3 | α | Mongan et al., 2002 | |||
| ACR-20 | DEG-3 | α | Mongan et al., 2002 | |||
| ACR-23 | DEG-3 | α | Mongan et al., 2002 | |||
| DEG-3 | DEG-3 | α | IL2, PVD, PVC, AVG, FLP, touch cells | deg-3 and des-2 in operon; better response to Ch than ACh | Treinin and Chalfie, 1995; Treinin et al., 1998; Yassin et al., 2001 | |
| DES-2 | DEG-3 | α | IL2, PVD, PVC, AVG, FLP, touch cells | =ACR-4; deg-3 and des-2 in operon; better response to Ch than ACh | Treinin et al., 1998 | |
| F11C7.1 | DEG-3 | Non-α | Predicted, no ESTs, no data | |||
| Y44A6E.1 | DEG-3 | Non-α | Several ESTs | |||
| Y73F8A.30 | DEG-3 | α | Predicted, no ESTs, no data | |||
| GAR-1 | GPCR | Sensory neurons in head, PVM | Lee et al., 1999; Lee et al., 2000 | |||
| GAR-2 | GPCR | Sensory neurons in head, ventral cord motor neurons, HSN | Lee et al., 2000; Fox et al., 2005 | |||
| GAR-3 | GPCR | Pharyngeal muscles, DA and/or SAB and/or I5 | =ACM-3 | Park et al., 2003; Steger and Avery, 2004; Fox et al., 2005 | ||
| ACC-1 | Ligand-gated chloride channel | Putrenko et al., 2005 | ||||
| ACC-2 | Ligand-gated chloride channel | Putrenko et al., 2005 | ||||
| ACC-3 | Ligand-gated chloride channel | Putrenko et al., 2005 | ||||
| ACC-4 | Ligand-gated chloride channel | Putrenko et al., 2005 | ||||
| aThe five groups of nicotinic AChR subunits are based on Mongan et al. (1998) and Jones and Sattelle (2004). | ||||||
| bThis is an incomplete list; it is likely that for many of these genes, there are additional expressing cells not listed. | ||||||
It is striking that the C. elegans genome is extraordinarily rich in genes encoding nicotinic-type receptor subunits (Mongan et al., 1998; Jones and Sattelle, 2004). Whereas mammals and birds have fewer than 20 AChR subunit genes, genetic and genomic analysis of C. elegans has thus far confirmed the transcription of 27 genes (Table 1). For some of these genes, heterologous expression individually and in combination (usually in Xenopus oocytes) has provided information about the properties of the receptor subunits; for other genes, the identity of the ligand is merely inferred. In addition, there are about 20 genes encoding putative ionotropic receptor subunits that have not been fully characterized, but could include additional AChR subunits.
Five classes of protein subunits have been identified, based on sequence similarity (see Table 1): UNC-29, UNC-38, ACR-8, ACR-16, and DEG-3 (Mongan et al., 1998; Mongan et al., 2002; Jones and Sattelle, 2004). Each class contains three to nine members, and some of the classes contain both α and non-α type subunits. The UNC-38 class contains three α subunits, and is most similar to insect a subunits. The UNC-29 class contains four non-α subunits; these proteins are similar to vertebrate skeletal muscle non-α and insect non-α subunits. The ACR-16 class consists of nine members (α and non-α), and its members resemble vertebrate α7 subunits. The ACR-8 class is α nematode specific grouping, and contains three α subunits. The DEG-3 class is also nematode specific, and consists of eight verified α and non-α subunits (there are also two genes, one α and one non-α, predicted to be in this class, but for which there are neither ESTs nor any expression data).
The best characterized C. elegans AChRs, based on electrophysiological recording and reconstitution in heterologous systems, are at neuromuscular junctions. The muscles of the body wall express two major types of ACh receptors: one type responds to levamisole and the other type responds to nicotine, but not levamisole (Richmond and Jorgensen, 1999). The levamisole-sensitive receptors on the body muscles appear to be heteromeric, and contain three essential subunits: UNC-29, UNC-38, and UNC-63 (Fleming et al., 1997; Richmond and Jorgensen, 1999; Culetto et al., 2004). LEV-1 and LEV-8 are also subunits, but they are either non-essential for the response to levamisole, or else they are only present in a subset of the receptors (Fleming et al., 1997; Towers et al., 2005). The nicotine-sensitive receptors appear to be homomeric, containing only the ACR-16 α subunit (Francis et al., 2005; Touroutine et al., 2005).
For about half of the AChR subunits, expression data from reporter studies and/or antibody localization data have been reported; in general, the different subunits appear to have complex and overlapping patterns of expression. This information is presented in Table 1.
As might be expected, loss-of-function mutations in any of the five subunits which comprise the muscle levamisole-sensitive receptor (described above) lead to levamisole resistance (Lewis et al., 1980b). However, loss-of-function mutations in unc-29, unc-38, and unc-63 confer strong levamisole resistance and a mild uncoordinated phenotype, while mutations in lev-1 and lev-8 lead to mild levamisole resistance and almost normal locomotion (Lewis et al., 1980b). In addition, acr-16 mutations, which eliminate the function of the muscle nicotine-sensitive receptor, have essentially wild-type locomotion (Francis et al., 2005; Touroutine et al., 2005). Thus, eliminating the function of either the levamisole-sensitive receptor or the nicotine-sensitive receptor does not have profound behavioral consequences. However, eliminating the function of both receptors (e.g., an unc-29; acr-16 double mutant or an unc-63;acr-16 double mutant) leads to a synthetic severe uncoordinated phenotype (Francis et al., 2005; Touroutine et al., 2005).
The deg-3 and des-2 genes are in an operon and encode similar AChR subunits (Treinin et al., 1998). There are rare dominant mutations in deg-3 that cause neuronal degeneration (presumably by hyperactivation of the channel); these mutations are suppressed by loss-of-function mutations in either des-2 or deg-3 (Treinin and Chalfie, 1995). The DEG-3 and DES-2 subunits can associate with each other to form a functional receptor in vitro (and presumably in vivo; Treinin et al., 1998). This heteromeric receptor responds more strongly to choline than acetylcholine (Yassin et al., 2001). The two genes are expressed in sensory neurons, and the DEG-3 protein is enriched in the sensory endings of these cells (Yassin et al., 2001). This suggested that the DEG-3/DES-2 receptor might play a role in sensory transduction, and deg-3 and des-2 mutants have been shown to be deficient in chemotaxis to choline (Yassin et al., 2001).
Two additional types of ACh receptors have been identified in C. elegans: G-protein coupled receptors (similar to vertebrate muscarinic receptors), and ligand-gated chloride channels (which are not found in vertebrates).
The apparent existence of muscarinic-type ACh receptors in C. elegans was first reported in 1983 (Culotti and Klein, 1983), and subsequently verified through analysis of the gar-1, gar-2, and gar-3 genes. The primary transcripts from all three of the gar genes undergo alternative splicing (Park et al., 2000; Suh et al., 2001; Park et al., 2003), leading to considerable protein diversity. Pharmacological profiles indicate that the GAR-3 isoforms are the most similar to “conventional” vertebrate muscarinic receptors, and bind the antagonist scopolamine and the agonist carbachol (Hwang et al., 1999; Park et al., 2003). The GAR-1 isoforms are somewhat different, and bind atropine, but not scopolamine (Lee et al., 1999), while GAR-2 binds neither atropine nor scopolamine (Lee et al., 2000). Complete expression profiles for these receptors have not been reported. However, GFP constructs indicated that gar-1 and gar-2 are expressed in neurons, including some sensory neurons in the head, PVM (gar-1 only), motor neurons in the ventral cord (gar-2), and HSN (gar-2) (Lee et al., 2000). gar-3 is expressed in pharyngeal muscle (Steger and Avery, 2004).
The four ACC proteins are most closely related to the C. elegans MOD-1 serotonin-gated chloride channel, and have no orthologs in vertebrate or Drosophila genomes (Putrenko et al., 2005). ACC-1 can form a homomeric ACh-gated chloride channel with an EC50 of 0.26 μM, or a heteromeric channel together with ACC-3, with an EC50 of 39.6 μM (Putrenko et al., 2005). ACC-2 can also form homomeric channels, with an EC50 of 9.54 μM. Nothing has yet been reported about the expression patterns of these proteins, or mutant phenotypes.
Several proteins have been described that seem to be associated with cholinergic function and are required for the expression, maturation, trafficking, and/or localization of particular ACh receptor subunits.
RIC-3 (a protein containing two transmembrane domains followed by three coiled-coil regions) is required for maturation of at least four types of ACh receptor: the EAT-2-containing pharyngeal AChR, the DEG-3/DES-2 neuronal AChR, the UNC-29-containing levamisole-sensitive muscle AChR, and the ACR-16-containing nicotine-sensitive muscle AChR (Halevi et al., 2002). RIC-3 is expressed in pharyngeal and body wall muscles, and in most or all neurons, and is localized to cell bodies. ric-3 mutants are Unc, Ric, and Lev (Nguyen et al., 1995; Miller et al., 1996).
LEV-10 (a transmembrane protein containing five extracellular CUB domains) is required for the clustering of post-synaptic UNC-29-containing AChRs (Gally et al., 2004). Consistent with this role, LEV-10 is expressed in body muscle, and is localized to post-synaptic cholinergic neuromuscular junctions (Gally et al., 2004).
EAT-18 (a small transmembrane protein) is somehow required for proper function of pharyngeal AChRs containing the EAT-2 non-α subunit, and apparently other pharyngeal nicotinic receptors as well (McKay et al., 2004). Surprisingly, the eat-18 gene and promoter are nested within the first intron of lev-10 (Gally et al., 2004). eat-18 mutants resemble eat-2 mutants, demonstrating the importance of EAT-18 for EAT-2 function (McKay et al., 2004).
CAM-1 (a Ror receptor tyrosine kinase) is required for the localization and clustering of ACR-16-containing nicotine-sensitive muscle AChRs (Francis et al., 2005), and also plays a role in the localization or function of the levamisole-sensitive receptors, but it is not involved in the localization or function of GABA receptors (Gottschalk et al., 2005). CAM-1 is expressed in muscles and is enriched in the muscle arms and especially at neuromuscular junctions. CAM-1 is also expressed in many neurons, including ventral cord cholinergic motor neurons. Analysis of cam-1 mutants shows that the protein is also involved in cell migration and axon guidance (Forrester et al., 1999).
Neither unc-50 nor unc-74 encodes an AChR subunit, yet mutations in these two genes lead to strong resistance to levamisole (Lewis et al., 1980b). Therefore, the UNC-50 and UNC-74 proteins must be required in some way for receptor response to levamisole (Lewis et al., 1980a). Although molecular analysis of neither unc-50 nor unc-74 has been reported directly, indirect reports indicate that the unc-50 gene encodes a novel type of transmembrane protein with identifiable mammalian and plant homologs.
There are also some proteins that clearly contribute to the stability, trafficking, and/or clustering of AChR subunits, but to a lesser extent than the gene products described above, and whose loss-of-function phenotypes are associated with partial reduction in the abundance of localized receptors. For example, NRA-1 is a copine localized to plasma membranes necessary for high-level expression of UNC-38 and LEV-1 (Gottschalk et al., 2005).
This is the most important acetylcholine-mediated behavior, involving by far the greatest number of cholinergic neurons. Locomotion (both crawling on surfaces and swimming in liquid) requires the ability to generate smooth, sinusoidal waves (at variable propagation rates) of either anterior-directed or posterior-directed body muscle contractions. The involvement of ACh in locomotion includes not only neuromuscular transmission, but also nerve-nerve transmission (which is poorly characterized). Thus, for example, ventral nerve cord cholinergic motor neurons express the ACR-2 and ACR-5 AChR subunits (Winnier et al., 1999; Hallam et al., 2000), while GABAergic motor neurons express LEV-8 (Towers et al., 2005). In addition, ACh appears to be involved in the regulation of the oscillator circuit controlling the rate of wave initiation: in addition to their uncoordinated movement, cholinergic (i.e., cha-1 and unc-17) mutants have a dramatically reduced rate of wave initiation.
Egg laying behavior involves the action of several neurotransmitters (see Egg-Laying), and ACh appears to affect egg laying through multiple mechanisms. The major effect of ACh appears to be an inhibition of egg laying, because animals deficient in cholinergic release (e.g., cha-1 and unc-17 mutants) are constitutive for egg-laying, and retain very few embryos (Bany et al., 2003). However, levamisole can stimulate egg laying (Kim et al., 2001), suggesting a possible stimulatory cholinergic mechanism operating through levamisole-sensitive receptors on muscles.
The egg-laying muscles are innervated by the HSN and VC cells; the VC cells also make synapses onto the HSNs (White et al., 1986). A plausible model is that ACh is released by VC cells onto the presynaptic terminals of the HSN cells, and interacts with inhibitory GAR-2 (and perhaps other) cholinergic receptors (Bany et al., 2003). This leads to decreased HSN release of serotonin. This model is complicated somewhat by the apparent multi-transmitter phenotype of both the HSNs and VC4/VC5 (Duerr et al., 2001).
The MC neuron is cholinergic and stimulates the pharynx muscle (via the EAT-2 receptor) to control pharyngeal pumping rate (McKay et al., 2004). Wild-type animals pump >250/min; pumping for animals ablated for MC is ~ 45/min (Avery and Horvitz, 1989; Raizen et al., 1995). It is likely that the slow growth rates of cha-1 and unc-17 mutants (Rand and Russell, 1984) are related to their reduced rate of pharyngeal pumping.
In addition, a number of other ACh receptor types are expressed on pharyngeal cells, and appear to mediate several aspects of pharynx function. For example, the muscarinic GAR-3 receptor, signaling through EGL-30 and GPB-2, is involved in control of pharyngeal muscle membrane potential (Steger and Avery, 2004).
Mutations in cha-1 (Thomas, 1990) and unc-17 (J. Rand, unpublished) cause dramatically longer and variable defecation cycle periods. However, when defecation does occur, it appears to be normal (Thomas, 1990). It is not known which cholinergic neurons are responsible for regulating this periodic behavior.
Several distinct cholinergic pathways in the male tail have been described that are involved in male mating behavior (see Male mating behavior). The PCB and PCC sensory neurons of the postcloacal sensilla are cholinergic, and together with the anal depressor and some still unidentified cells, mediate periodic contractions (“prodding” behavior) of the protractor muscles (Liu and Sternberg, 1995; Garcia et al., 2001). The subsequent spicule insertion requires prolonged contraction of the protractor muscles, mediated by the cholinergic SPC motor neurons. Protractor contraction (both the periodic prodding and the prolonged contraction) is severely deficient in cha-1 mutants, and is potentiated in wild-type animals by aldicarb; the cholinergic signaling is mediated by multiple receptors, including distinct levamisole-, nicotine-, and arecoline-sensitive receptors (Garcia et al., 2001).
Although we are used to thinking that each neuron releases a single neurotransmitter, this turns out to be an oversimplification. For example, the four motor neurons with significant output to the egg-laying muscles, HSNL/R and VC4/5 appear to release both ACh and serotonin (Duerr et al., 2001). It is not known if the two transmitters are colocalized in the same vesicles, or if they are released from the same synapses.
One of the consequences of starvation is the triggering of protein degradation in muscles. This may be monitored as the disappearance of muscle reporter expression in response to starvation (Zdinak et al., 1997). The starvation-dependent proteolysis is negatively regulated by cholinergic input to the muscles (Szewczyk et al., 2000). For example, mutants deficient in ACh release or reception (e.g., cha-1, unc-17, unc-13, unc-38) have enhanced starvation-induced protein turnover, while the cholinergic agonist levamisole completely prevents starvation-induced turnover in muscle cells (Szewczyk et al., 2000).
Ruaud and Bessereau (2006) have reported that application of the nicotinic receptor agonist DMPP slowed overall C. elegans development during the L2 stage but did not affect the molt timing, so that the L2/L3 molt occurred before the L3 cuticle was ready, and the animals died. This suggests the uncoupling of a “developmental timer” apparently regulated by cholinergic signaling from a “molting timer”. The action of the drug was not due to general toxicity, but rather was specific and was mediated through UNC-63-containing receptors, because unc-63 mutants were partially resistant to the stage-specific lethality of DMPP. However, the DMPP sensitivity was not mediated by the muscle levamisole-sensitive receptors, but rather by neuronal UNC-63-containing receptors. The specific neuronal pathways mediating this effect have not yet been determined.
The author's research has been supported by grants from the National Institute of General Medical Sciences.
Alfonso, A., Grundahl, K., Duerr, J.S., Han, H.-P., and Rand, J.B. (1993). The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science 261, 617–619. Abstract Article
Alfonso, A., Grundahl, K., McManus, J.R., Asbury, J.M., and Rand, J.B. (1994a). Alternative splicing leads to two cholinergic proteins in Caenorhabditis elegans. J. Mol. Biol. 241, 627–630. Abstract Article
Alfonso, A., Grundahl, K., McManus, J.R., and Rand, J.B. (1994b). Cloning and characterization of the choline acetyltransferase structural gene (cha-1) from C. elegans. J. Neurosci. 14, 2290–2300. Abstract
Apparsundaram, S., Ferguson, S.M., George, A.L., Jr., and Blakely, R.D. (2000). Molecular cloning of a human, hemicholinium-3-sensitive choline transporter. Biochem. Biophys. Res. Commun. 276, 862–867. Abstract Article
Arpagaus, M., Fedon, Y., Cousin, X., Chatonnet, A., Bergé, J.-B., Fournier, D., and Toutant, J.-P. (1994). cDNA sequence, gene structure, and in vitro expression of ace-1, the gene encoding acetylcholinesterase of class A in the nematode Caenorhabditis elegans. J. Biol. Chem. 269, 9957–9965. Abstract
Avery, L. and Horvitz, H.R. (1989). Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron 3, 473–485. Abstract Article
Ballivet, M., Alliod, C., Bertrand, S., and Bertrand, D. (1996). Nicotinic acetylcholine receptors in the nematode Caenorhabditis elegans. J. Mol. Biol. 258, 261–269. Article
Bany, I.A., Dong, M.Q., and Koelle, M.R. (2003). Genetic and cellular basis for acetylcholine inhibition of Caenorhabditis elegans egg-laying behavior. J. Neurosci. 23, 8060–8069. Abstract
Baylis, H.A., Matsuda, K., Squire, M.D., Fleming, J.T., Harvey, R.J., Darlison, M.G., Barnard, E.A., and Sattelle, D.B. (1997). ACR-3, a Caenorhabditis elegans nicotinic acetylcholine receptor subunit. Molecular cloning and functional expression. Recept. Channels 5, 149–158.
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94. Abstract
Bussiere, M., Vance, J.E., Campenot, R.B., and Vance, D.E. (2001). Compartmentalization of choline and acetylcholine metabolism in cultured sympathetic neurons. J. Biochem. (Tokyo) 130, 561–568. Abstract
Collier, B. (1988). About the coupling of acetylcholine hydrolysis and choline uptake at cholinergic nerve terminals. J. Neurochem. 50, 323–324. Abstract Article
Combes, D., Fedon, Y., Grauso, M., Toutant, J.P., and Arpagaus, M. (2000). Four genes encode acetylcholinesterases in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae. cDNA sequences, Genomic structures, mutations and in vivo expression. J. Mol. Biol. 300, 727–742. Abstract Article
Combes, D., Fedon, Y., Toutant, J.P., and Arpagaus, M. (2003). Multiple ace genes encoding acetylcholinesterases of Caenorhabditis elegans have distinct tissue expression. Eur. J. Neurosci. 18, 497–512. Abstract Article
Culetto, E., Baylis, H.A., Richmond, J.E., Jones, A.K., Fleming, J.T., Squire, M.D., Lewis, J.A., and Sattelle, D.B. (2004). The Caenorhabditis elegans unc-63 gene encodes a levamisole-sensitive nicotinic acetylcholine receptor β subunit. J. Biol. Chem. 279, 42476–42483. Abstract Article
Culetto, E., Combes, D., Fedon, Y., Roig, A., Toutant, J.P., and Arpagaus, M. (1999). Structure and promoter activity of the 5′ flanking region of ace-1, the gene encoding acetylcholinesterase of Class A in Caenorhabditis elegans. J. Mol. Biol. 290, 951–966. Abstract Article
Culotti, J.G. and Klein, W.L. (1983). Occurrence of muscarinic acetylcholine receptors in wild-type and cholinergic mutants of C. elegans. J. Neurosci. 3, 359–368. Abstract
Culotti, J.G., von Ehrenstein, G., Culotti, M.R., and Russell, R.L. (1981). A second class of acetylcholinesterase-deficient mutants of the nematode Caenorhabditis elegans. Genetics 97, 281–305. Abstract
del Castillo, J., de Mello, W.C., and Morales, T. (1963). The physiological role of acetylcholine in the neuromuscular system of Ascaris lumbricoides. Arch. Int. Physiol. Biochim. 71, 741–757. Abstract
del Castillo, J., de Mello, W.C., and Morales, T. (1967). The initiation of action potentials in the somatic musculature of Ascaris lumbricoides. J. Exp. Biol. 46, 263–279. Abstract
Duerr, J.S., Gaskin, J., and Rand, J.B. (2001). Identified neurons in C. elegans co-express vesicular transporters for acetylcholine and monoamines. Am. J. Physiol. Cell Physiol. 280, C1616–C1622. Abstract
Eiden, L.E. (1998). The cholinergic gene locus. J. Neurochem. 70, 2227–2240. Abstract
Erickson, J.D., Varoqui, H., Schäfer, M.K.H., Modi, W., Diebler, M.-F., Weihe, E., Rand, J., Eiden, L.E., Bonner, T.I., and Usdin, T.B. (1994). Functional identification of a vesicular acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem. 269, 21929–21932. Abstract
Ferguson, S.M., Savchenko, V., Apparsundaram, S., Zwick, M., Wright, J., Heilman, C.J., Yi, H., Levey, A.I., and Blakely, R.D. (2003). Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J. Neurosci. 23, 9697–9709. Abstract
Fleming, J.T., Squire, M.D., Barnes, T.M., Tornoe, C., Matsuda, K., Ahnn, J., Fire, A., Sulston, J.E., Barnard, E.A., Sattelle, D.B., and Lewis, J.A. (1997). Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits. J. Neurosci. 17, 5843–5857. Abstract
Forrester, W.C., Dell, M., Perens, E., and Garriga, G. (1999). A C. elegans Ror receptor tyrosine kinase regulates cell motility and asymmetric cell division. Nature 400, 881–885. Abstract Article
Fox, R.M., Von Stetina, S.E., Barlow, S.J., Shaffer, C., Olszewski, K.L., Moore, J.H., Dupuy, D., Vidal, M., and Miller, D.M., III (2005). A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6, 42. Article
Francis, M.M., Evans, S.P., Jensen, M., Madsen, D.M., Mancuso, J., Norman, K.R., and Maricq, A.V. (2005). The Ror receptor tyrosine kinase CAM-1 is required for ACR-16-mediated synaptic transmission at the C. elegans neuromuscular junction. Neuron 46, 581–594. Abstract Article
Friedrich, A., George, R.L., Bridges, C.C., Prasad, P.D., and Ganapathy, V. (2001). Transport of choline and its relationship to the expression of the organic cation transporters in a rat brain microvessel endothelial cell line (RBE4). Biochim. Biophys. Acta 1512, 299–307. Abstract Article
Gally, C., Eimer, S., Richmond, J.E., and Bessereau, J.L. (2004). A transmembrane protein required for acetylcholine receptor clustering in Caenorhabditis elegans. Nature 431, 578–582. Abstract Article
Garcia, L.R., Mehta, P., and Sternberg, P.W. (2001). Regulation of distinct muscle behaviors controls the C. elegans male's copulatory spicules during mating. Cell 107, 777–788. Abstract Article
Gottschalk, A., Almedom, R.B., Schedletzky, T., Anderson, S.D., Yates, J.R., III, and Schafer, W.R. (2005). Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis elegans. EMBO J. 24, 2566–2578. Abstract Article
Grauso, M., Culetto, E., Combes, D., Fedon, Y., Toutant, J.P., and Arpagaus, M. (1998). Existence of four acetylcholinesterase genes in the nematodes Caenorhabditis elegans and Caenorhabditis briggsae. FEBS Lett. 424, 279–284. Abstract Article
Halevi, S., McKay, J., Palfreyman, M., Yassin, L., Eshel, M., Jorgensen, E., and Treinin, M. (2002). The C. elegans ric-3 gene is required for maturation of nicotinic acetylcholine receptors. EMBO J. 21, 1012–1020. Abstract Article
Hallam, S., Singer, E., Waring, D., and Jin, Y.S. (2000). The C. elegans NeuroD homolog cnd-1 functions in multiple aspects of motor neuron fate specification. Development 127, 4239–4252. Abstract
Herman, R.K. and Kari, C.K. (1985). Muscle-specific expression of a gene affecting acetylcholinesterase in the nematode Caenorhabditis elegans. Cell 40, 509–514. Abstract Article
Hosono, R., Kuno, S., and Midsukami, M. (1985). Temperature-sensitive mutations causing reversible paralysis in Caenorhabditis elegans. J. Exp. Zool. 235, 409–421. Abstract Article
Hosono, R., Sassa, T., and Kuno, S. (1989). Spontaneous mutations of trichlorfon resistance in the nematode Caenorhabditis elegans. Zool. Sci. 6, 697–708.
Hwang, J.M., Chang, D.J., Kim, U.S., Lee, Y.S., Park, Y.S., Kaang, B.K., and Cho, N.J. (1999). Cloning and functional characterization of a Caenorhabditis elegans muscarinic acetylcholine receptor. Recept. Channels 6, 415–424. Abstract
Johnson, C.D., Duckett, J.G., Culotti, J.G., Herman, R.K., Meneely, P.M., and Russell, R.L. (1981). An acetylcholinesterase-deficient mutant of the nematode Caenorhabditis elegans. Genetics 97, 261–279. Abstract
Johnson, C.D., Rand, J.B., Herman, R.K., Stern, B.D., and Russell, R.L. (1988). The acetylcholinesterase genes of C. elegans: identification of a third gene (ace-3) and mosaic mapping of a synthetic lethal phenotype. Neuron 1, 165–173. Abstract Article
Johnson, C.D. and Russell, R.L. (1983). Multiple molecular forms of acetylcholinesterase in the nematode Caenorhabditis elegans. J. Neurochem. 41, 30–46. Abstract Article
Jones, A.K. and Sattelle, D.B. (2004). Functional genomics of the nicotinic acetylcholine receptor gene family of the nematode, Caenorhabditis elegans. BioEssays 26, 39–49. Abstract Article
Jope, R.S. and Jenden, D.J. (1980). The utilization of choline and acetyl coenzyme A for the synthesis of acetylcholine. J. Neurochem. 35, 318–325. Abstract Article
Kim, H., Rogers, M.J., Richmond, J.E., and McIntire, S.L. (2004). SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature 430, 891–896. Abstract Article
Kim, J., Poole, D.S., Waggoner, L.E., Kempf, A., Ramirez, D.S., Treschow, P.A., and Schafer, W.R. (2001). Genes affecting the activity of nicotinic receptors involved in Caenorhabditis elegans egg-laying behavior. Genetics 157, 1599–1610. Abstract
Kitamoto, T., Wang, W., and Salvaterra, P.M. (1998). Structure and organization of the Drosophila cholinergic locus. J. Biol. Chem. 273, 2706–2713. Abstract Article
Kolson, D.L. and Russell, R.L. (1985). A novel class of acetylcholinesterase, revealed by mutations, in the nematode Caenorhabditis elegans. J. Neurogenet. 2, 93–110. Abstract
Lackner, M.R., Nurrish, S.J., and Kaplan, J.M. (1999). Facilitation of synaptic transmission by EGL-30 Gqα and EGL-8 PLCβ: DAG binding to UNC-13 is required to stimulate acetylcholine release. Neuron 24, 335–346. Abstract Article
Lee, Y.S., Park, Y.S., Chang, D.J., Hwang, J.M., Min, C.K., Kaang, B.K., and Cho, N.J. (1999). Cloning and expression of a G protein-linked acetylcholine receptor from Caenorhabditis elegans. J. Neurochem. 72, 58–65. Abstract Article
Lee, Y.S., Park, Y.S., Nam, Y., Suh, S.J., Lee, F., Kaang, B.K., and Cho, N.J. (2000). Characterization of GAR-2, a novel G protein-linked acetylcholine receptor from Caenorhabditis elegans. J. Neurochem. 75, 1800–1809. Abstract Article
Lewis, J.A., Wu, C.H., Levine, J.H., and Berg, H. (1980a). Levamisole-resistant mutants of the nematode Caenorhabditis elegans appear to lack pharmacological acetylcholine receptors. Neuroscience 5, 967–989. Abstract Article
Lewis, J.A., Wu, C.-H., Berg, H., and Levine, J.H. (1980b). The genetics of levamisole resistance in the nematode Caenorhabditis elegans. Genetics 95, 905–928. Abstract
Liu, K.S. and Sternberg, P.W. (1995). Sensory regulation of male mating behavior in Caenorhabditis elegans. Neuron 14, 79–89. Abstract Article
Loewi, O. (1921). Über humerole übertragbarkeit der herznervenwirkung. I. Mitteilung. Pflugers Arch. 189, 239–242. Article
Matthies, D.S., Fleming, P.A., Wilkes, D.M., and Blakely, R.D. (2006). The Caenorhabditis elegans choline transporter CHO-1 sustains acetylcholine synthesis and motor function in an activity-dependent manner. J. Neurosci. 26, 6200–6212. Abstract Article
McKay, J.P., Raizen, D.M., Gottschalk, A., Schafer, W.R., and Avery, L. (2004). eat-2 and eat-18 are required for nicotinic neurotransmission in the Caenorhabditis elegans pharynx. Genetics 166, 161–169. Abstract Article
Mellanby, H. (1955). The identification and estimation of acetylcholine in three parasitic nematodes (Ascaris lumbricoides, Litomosoides carinii, and the microfilariae of Dirofilaria repens). Parasitology 45, 287–294. Abstract
Miller, K.G., Alfonso, A., Nguyen, M., Crowell, J.A., Johnson, C.D., and Rand, J.B. (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. U.S.A. 93, 12593–12598. Abstract Article
Miller, K.G., Emerson, M.D., McManus, J.R., and Rand, J.B. (2000). RIC-8 (synembryn): A novel conserved protein that is required for Gqα signaling in the C. elegans nervous system. Neuron 27, 289–299. Abstract Article
Miller, K.G., Emerson, M.D., and Rand, J.B. (1999). Goα and diacylglycerol kinase negatively regulate the Gqα pathway in C. elegans. Neuron 24, 323–333. Abstract Article
Miller, K.G. and Rand, J.B. (2000). A role for RIC-8 (Synembryn) and GOA-1 (Goα) in regulating a subset of centrosome movements during early embryogenesis in Caenorhabditis elegans. Genetics 156, 1649–1660.
Mongan, N.P., Baylis, H.A., Adcock, C., Smith, G.R., Sansom, M.S., and Sattelle, D.B. (1998). An extensive and diverse gene family of nicotinic acetylcholine receptor alpha subunits in Caenorhabditis elegans. Recept. Channels 6, 213–228. Abstract
Mongan, N.P., Jones, A.K., Smith, G.R., Sansom, M.S.P., and Sattelle, D.B. (2002). Novel α7-like nicotinic acetylcholine receptor subunits in the nematode Caenorhabditis elegans. Protein Sci. 11, 1162–1171. Abstract Article
Nguyen, M., Alfonso, A., Johnson, C.D., and Rand, J.B. (1995). Caenorhabditis elegans mutants resistant to inhibitors of acetylcholinesterase. Genetics 140, 527–535. Abstract
Nonet, M.L., Grundahl, K., Meyer, B.J., and Rand, J.B. (1993). Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73, 1291–1305. Abstract Article
Nonet, M.L., Saifee, O., Zhao, H., Rand, J.B., and Wei, L. (1998). Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J. Neurosci. 18, 70–80. Abstract
Nurrish, S., Ségalat, L., and Kaplan, J.M. (1999). Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron 24, 231–242. Abstract Article
O'Regan, S., Traiffort, E., Ruat, M., Cha, N., Compaoré, D., and Meunier, F.M. (2000). An electric lobe suppressor for a yeast choline transport mutation belongs to a new family of transporter-like proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1835–1840. Abstract Article
Okuda, T., Haga, T., Kanai, Y., Endou, H., Ishihara, T., and Katsura, I. (2000). Identification and characterization of the high-affinity choline transporter. Nat. Neurosci. 3, 120–125. Abstract Article
Palavalli, L.H., Brendza, K.M., Haakenson, W., Cahoon, R.E., McLaird, M., Hicks, L.M., McCarter, J.P., Williams, D.J., Hresko, M.C., and Jez, J.M. (2006). Defining the role of phosphomethylethanolamine N-methyltransferase from Caenorhabditis elegans in phosphocholine biosynthesis by biochemical and kinetic analysis. Biochemistry 45, 6056–6065. Abstract Article
Park, Y.S., Kim, S., Shin, Y., Choi, B., and Cho, N.J. (2003). Alternative splicing of the muscarinic acetylcholine receptor GAR-3 in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 308, 961–965. Abstract Article
Park, Y.S., Lee, Y.S., Cho, N.J., and Kaang, B.K. (2000). Alternative splicing of gar-1, a Caenorhabditis elegans G-protein-linked acetylcholine receptor gene. Biochem. Biophys. Res. Commun. 268, 354–358. Abstract Article
Putrenko, I., Zakikhani, M., and Dent, J.A. (2005). A family of acetylcholine-gated chloride channel subunits in Caenorhabditis elegans. J. Biol. Chem. 280, 6392–6398. Abstract Article
Raizen, D.M., Lee, R.Y., and Avery, L. (1995). Interacting genes required for pharyngeal excitation by motor neuron MC in Caenorhabditis elegans. Genetics 141, 1365–1382. Abstract
Rand, J.B. (1989). Genetic analysis of the cha-1 - unc-17 gene complex in Caenorhabditis. Genetics 122, 73–80. Abstract
Rand, J.B. and Nonet, M.L. (1997). Neurotransmitter assignments for specific neurons. In C. elegans II, D.L. Riddle, T. Blumenthal, B.J. Meyer, and J.R. Preiss, eds. (Cold Spring Harbor Laboratory Press), pp. 1049–1052.
Rand, J.B. and Russell, R.L. (1984). Choline acetyltransferase-deficient mutants of the nematode Caenorhabditis elegans. Genetics 106, 227–248. Abstract
Rand, J.B. and Russell, R.L. (1985). Properties and partial purification of choline acetyltransferase from the nematode Caenorhabditis elegans. J. Neurochem. 44, 189–200. Abstract Article
Reynolds, N.K., Schade, M.A., and Miller, K.G. (2005). Convergent, RIC-8-dependent Gα signaling pathways in the Caenorhabditis elegans synaptic signaling network. Genetics 169, 651–670. Abstract Article
Richmond, J.E. and Jorgensen, E.M. (1999). One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat. Neurosci. 2, 791–797. Abstract Article
Ruaud, A.F. and Bessereau, J.L. (2006). Activation of nicotinic receptors uncouples a developmental timer from the molting timer in C. elegans. Development 133, 2211–2222. Abstract Article
Schade, M.A., Reynolds, N.K., Dollins, C.M., and Miller, K.G. (2005). Mutations that rescue the paralysis of Caenorhabditis elegans ric-8 (synembryn) mutants activate the Gαs pathway and define a third major branch of the synaptic signaling network. Genetics 169, 631–649. Abstract Article
Selkirk, M.E., Lazari, O., Hussein, A.S., and Matthews, J.B. (2005). Nematode acetylcholinesterases are encoded by multiple genes and perform non-overlapping functions. Chem. Biol. Interact. 157–158, 263–268. Article
Sieburth, D., Ch'ng, Q., Dybbs, M., Tavazoie, M., Kennedy, S., Wang, D., Dupuy, D., Rual, J.F., Hill, D.E., Vidal, M., Ruvkun, G., and Kaplan, J.M. (2005). Systematic analysis of genes required for synapse structure and function. Nature 436, 510–517. Abstract Article
Squire, M.D., Tornoe, C., Baylis, H.A., Fleming, J.T., Barnard, E.A., and Satelle, D.B. (1995). Molecular cloning and functional co-expression of a Caenorhabditis elegans nicotinic acetylcholine receptor subunit (acr-2). Recept. Channels 3, 107–115.
Steger, K.A. and Avery, L. (2004). The GAR-3 muscarinic receptor cooperates with calcium signals to regulate muscle contraction in the Caenorhabditis elegans pharynx. Genetics 167, 633–643. Abstract Article
Stern, B.D. (1986). Acetylcholinesterase from Caenorhabditis elegans: Partial purification and immunocytochemistry of Class C, and discovery of Class D.Ph.D. Thesis, University of Pittsburgh.
Suh, S., Park, Y.S., Lee, Y.S., Cho, T.J., Kaang, B.K., and Cho, N.J. (2001). Three functional isoforms of GAR-2, a Caenorhabditis elegans G-protein-linked acetylcholine receptor, are produced by alternative splicing. Biochem. Biophys. Res. Commun. 288, 1238–1243. Abstract Article
Szewczyk, N.J., Hartman, J.J., Barmada, S.J., and Jacobson, L.A. (2000). Genetic detects in acetylcholine signalling promote protein degradation in muscle cells of Caenorhabditis elegans. J. Cell Sci. 113, 2003–2010. Abstract
Thomas, J.H. (1990). Genetic analysis of defecation in Caenorhabditis elegans. Genetics 124, 855–872. Abstract
Touroutine, D., Fox, R.M., Von Stetina, S.E., Burdina, A., Miller, D.M., III, and Richmond, J.E. (2005). acr-16 encodes an essential subunit of the levamisole-resistant nicotinic receptor at the Caenorhabditis elegans neuromuscular junction. J. Biol. Chem. 280, 27013–27021. Abstract Article
Towers, P.R., Edwards, B., Richmond, J.E., and Sattelle, D.B. (2005). The Caenorhabditis elegans lev-8 gene encodes a novel type of nicotinic acetylcholine receptor α subunit. J. Neurochem. 93, 1–9. Abstract Article
Treinin, M. and Chalfie, M. (1995). A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 14, 871–877. Abstract Article
Treinin, M., Gillo, B., Liebman, L., and Chalfie, M. (1998). Two functionally dependent acetylcholine subunits are encoded in a single Caenorhabditis elegans operon. Proc. Natl. Acad. Sci. U.S.A. 95, 15492–15495. Abstract Article
Wang, Y., Cao, Z., Newkirk, R.F., Ivy, M.T., and Townsel, J.G. (2001). Molecular cloning of a cDNA for a putative choline co-transporter from Limulus CNS. Gene 268, 123–131. Abstract Article
White, J.G., Southgate, E., Thomson, J.N., and Brenner, S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Phil. Trans. R. Soc. Lond. B 314, 1–340. Abstract
Winnier, A.R., Meir, J.Y.J., Ross, J.M., Tavernarakis, N., Driscoll, M., Ishihara, T., Katsura, I., and Miller, D.M., III (1999). UNC-4/UNC-37-dependent repression of motor neuron-specific genes controls synaptic choice in Caenorhabditis elegans. Genes Dev. 13, 2774–2786. Abstract Article
Wu, X., Fei, Y.J., Huang, W., Chancy, C., Leibach, F.H., and Ganapathy, V. (1999). Identity of the F52F12.1 gene product in Caenorhabditis elegans as an organic cation transporter. Biochim. Biophys. Acta Bio-Membr. 1418, 239–244. Abstract Article
Yassin, L., Gillo, B., Kahan, T., Halevi, S., Eshel, M., and Treinin, M. (2001). Characterization of the DEG-3/DES-2 receptor: A nicotinic acetylcholine receptor that mutates to cause neuronal degeneration. Mol. Cell. Neurosci. 17, 589–599. Abstract Article
Yook, K.J., Proulx, S.R., and Jorgensen, E.M. (2001). Rules of nonallelic noncomplementation at the synapse in Caenorhabditis elegans. Genetics 158, 209–220. Abstract
Zdinak, L.A., Greenberg, I.B., Szewczyk, N.J., Barmada, S.J., Cardamone-Rayner, M., Hartman, J.J., and Jacobson, L.A. (1997). Transgene-coded chimeric proteins as reporters of intracellular proteolysis: Starvation-induced catabolism of a lacZ fusion protein in muscle cells of Caenorhabditis elegans. J. Cell. Biochem. 67, 143–153. Abstract
Zhao, H. and Nonet, M.L. (2000). A retrograde signal is involved in activity-dependent remodeling at a C. elegans neuromuscular junction. Development 127, 1253–1266. Abstract
*Edited by Erik M. Jorgensen and Joshua M. Kaplan. Last revised December 5, 2006. Published January 30, 2007. This chapter should be cited as: Rand, J.B. Acetylcholine (January 30, 2007), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.131.1, http://www.wormbook.org.
Copyright: © 2007 James B. Rand. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Tel: 405 271-7681. E-mail: James-Rand@omrf.ouhsc.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.