Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
The lack of robust functional genomic tools for the majority of parasitic nematodes severely hampers functional genomic study of these organisms. However, some progress has been made in the area of transgenesis for two important groups of parasites: Strongyloides and related genera, representing soil-transmitted intestinal parasites, and Brugia malayi, representing the mosquito-transmitted lymphatic filariae. Gonadal microinjection suffices for gene transfer into Strongyloides and Parastrongyloides owing to the presence of one or more generations of free-living females, with similar body plans to C. elegans hermaphrodites in their life cycles. It has been possible to generate transiently transgenic parasites by gonadal microinjection of plasmid vectors into Strongyloides and Parastrongyloides since 2006. More recently, chromosomal integration of microinjected plasmid vectors using the piggyBac transposon system has enabled derivation of the first stable transgenic lines of Strongyloides ratti. Protocols for establishing and cryopreserving stable transgenic lines of S. ratti are included in this chapter. Gonadal microinjection is impractical for other parasitic nematodes, such as the filariae, which lack free-living adults in their life cycles. Until recently, biolistic transfer of DNA coated microparticles has been the primary method of gene transfer in the filariae, allowing analysis of functional aspects such as core promoter domains and cis-acting elements affecting trans-splicing in B. malayi. Biolistic DNA transfer failed, however, to yield transgenic parasites that were sufficiently viable to be returned to the host for continuous passage. By contrast, chemically mediated gene transfer, involving incubation of developing larvae with DNA/calcium co-precipitates, yields fully viable transgenic larvae of B. malayi that can be maintained in laboratory hosts. This represents a major advancement for B. malayi and promises to be a method that is widely applicable to the majority of parasitic nematodes that lack a free-living generation. This chapter contains protocols for biolistic and chemically mediated transfection of B. malayi.
Parasitic nematodes exact a staggering toll in human suffering, causing debilitating or potentially blinding disease in roughly one fifth of the human population (Brooker, 2010; Hotez et al., 2009). Parasitic nematodes also have a profound negative impact on domestic animals, causing significant economic losses to agriculture in developed economies (Charlier et al., 2009) and negatively impacting food security in the developing world (Krecek and Waller, 2006).
From a highly simplified viewpoint, the medically important parasitic nematodes may be divided into those worms dwelling in the gastrointestinal tracts of their vertebrate hosts and those inhabiting the lymphatics or connective tissues of their hosts. Familiar examples of the former group include the ascarid roundworms, the hookworms and the threadworms.
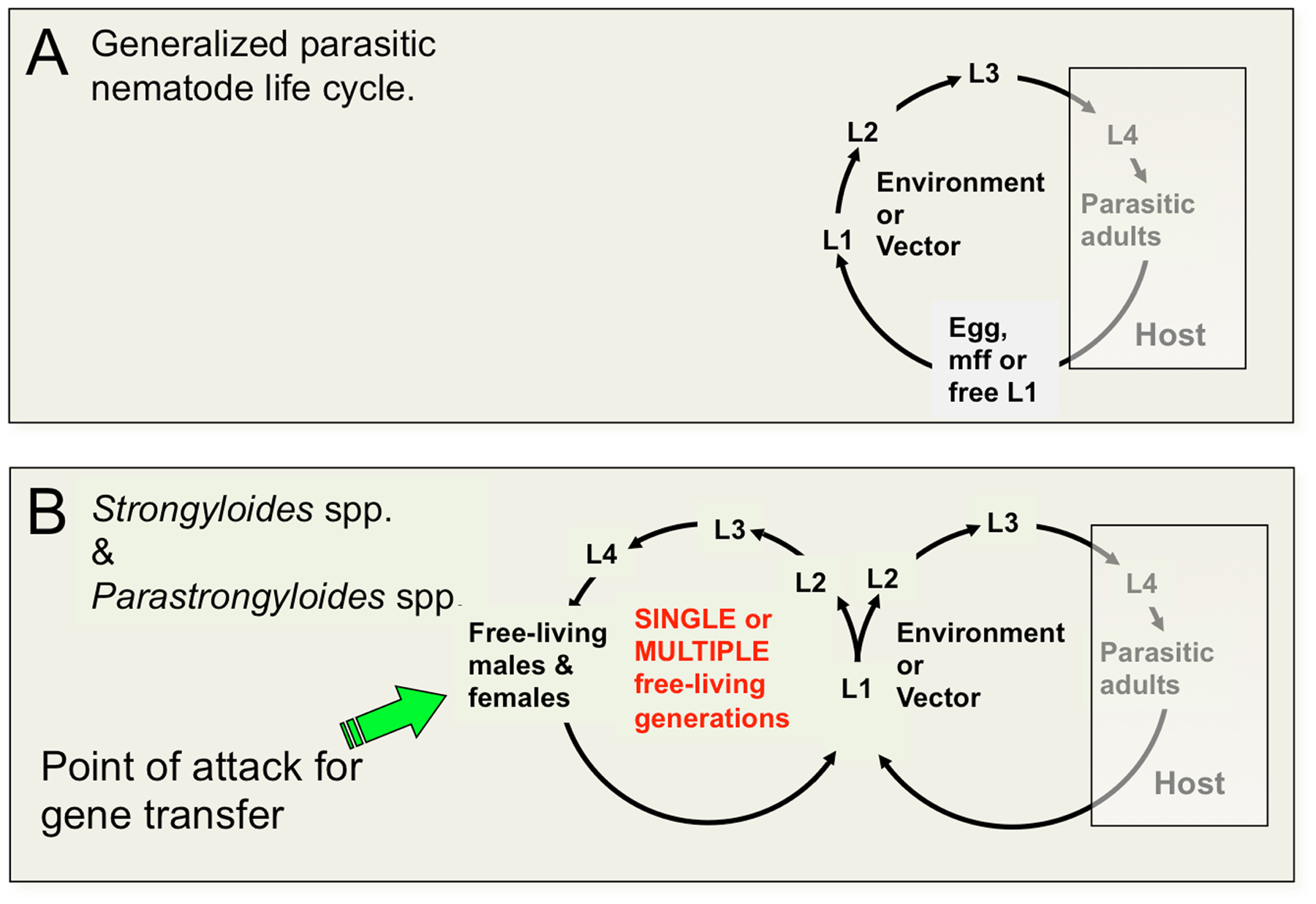
The threadworms include members of the genus Strongyloides, which is notable among the vertebrate animal parasites in being the only group that is capable of executing one or more complete generations of development outside the vertebrate host (Figure 1). The ability of Strongyloides spp. and related genera to undertake alternate generations of free-living development has contributed to their being adopted as models for basic molecular, cellular and developmental biological research. The basic biology and medical significance of the genus Strongyloides is described elsewhere in WormBook (see Strongyloides spp.). Life cycles of most parasitic nematodes involve an invariant pattern of development in which eggs, microfilariae or first-stage larvae (L1) are released from the host in the feces or acquired during blood feeding by an arthropod vector, followed by a series of larval stages in the environment extrinsic to the definitive host. Extrinsic development culminates in the formation of a developmentally arrested larval stage that acts as the infectious stage, which invades the vertebrate host (Figure 1A). The life cycles of Strongyloides spp. and related genera are unique among parasitic nematodes in involving one or more generations of free-living development followed by formation of infective third-stage larvae and then a generation of parasitic development (Figure 1B).
|
Figure 1. Life cycles of animal parasitic nematodes generally and of Strongyloides spp. and related parasites compared. A Generalized parasitic nematode life cycle featuring invariant pattern of development of pre-infective to infective larvae outside the host and development to reproductive adults and their progeny in the host. Status of the L3 as the infective stage is common to vertebrate animal parasites in nematode clades III, IV and V. Animal parasites in Clade I, principally Trichinella and Trichuris, infect the host as L1. B. Life cycle of Strongyloides and related genera featuring one or more generations of free-living development. The free-living female constitutes an advantageous target for gene transfer by gonadal microinjection.
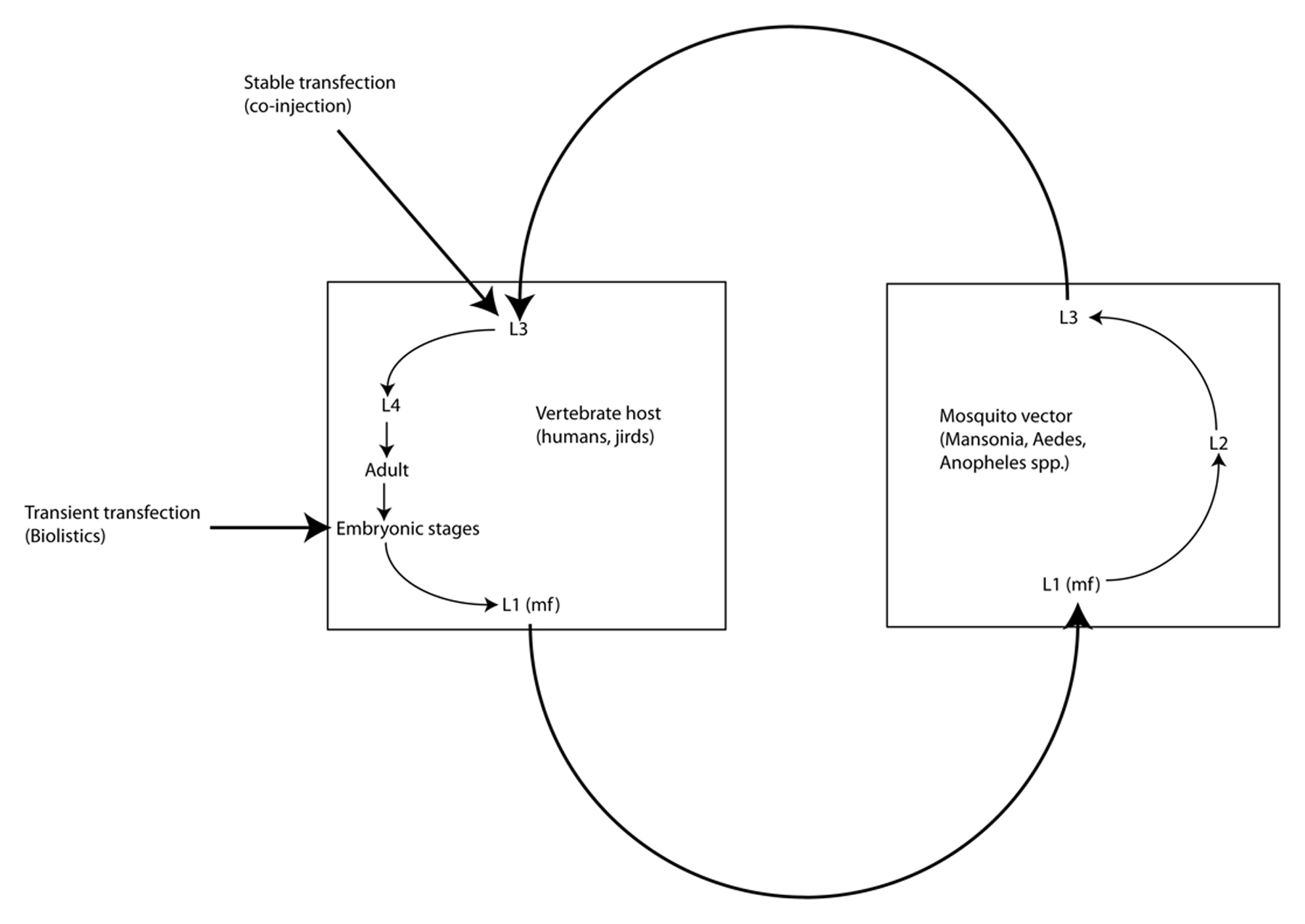
Brugia malayi is one of eight filarial parasites that infect humans (the others being Brugia timori, Wuchereria bancrofti, Onchocerca volvulus, Loa loa, Mansonella streptocerca, M. perstans and M. ozzardi) (Liu et al., 2010; Walther and Muller, 2003). In contrast to the gastrointestinal nematodes, filariae are parasites of the tissues and tissue spaces of their vertebrate hosts. Adult B. malayi, B. timori, and W. bancrofti live in the lymphatics and cause elephantiasis in humans; L. loa, M. streptocerca and O. volvulus (the cause of river blindness, or onchocerciasis) inhabit the skin and connective tissues; and M. perstans and M. ozzardi inhabit the abdominal cavity. Female filariae are ovoviviparous and release live microfilariae into the surrounding host tissues: lymph and eventually blood in the case of the lymphatic dwelling filariae, L. loa and Mansonella spp., and skin in the case of O. volvulus. These microfilariae (MF), which are essentially pre-first-stage larvae, are taken up during blood feeding by arthropod vectors (mosquitoes in the case of the lymphatic dwelling filariae), which support development from MF to L1 and then from L1-L3, The infective L3 (L3i) are then transmitted to recipient hosts during another blood feeding (Figure 2). All filariae are obligate parasites, meaning that they cannot be cultured outside of a host for any period of time. B. malayi is unique among the human filarial parasites in that it is the only one of the four that can be cultured in a laboratory animal host (Ash and Riley, 1970); the others are obligate parasites of humans. This has made B. malayi the model of choice for the study of the human filariae.
|
Figure 2. Life cycle of Brugia spp. Filariae such as Brugia spp. live as adults in the blood or lymphatic circulation, the connective tissues or tissue spaces. They release microfilariae (mf) into surrounding tissues where they are ingested by hematophagous arthropod vectors (mosquitoes in the case of Brugia spp.) in the process of blood feeding. Because they are obligate parasites, propagation of stable transgenic lines of filariae will require passage through both hosts and vectors. Embryos within the uteri of female worms explanted from laboratory animal hosts (gerbils) constitute practical targets for biolistic transfer of DNA constructs. Co-injection of infective L3 from mosquitoes with DNA constructs in the presence of high Ca++ into the peritoneal cavities of gerbils has resulted in heritable transformation of Brugia malayi.
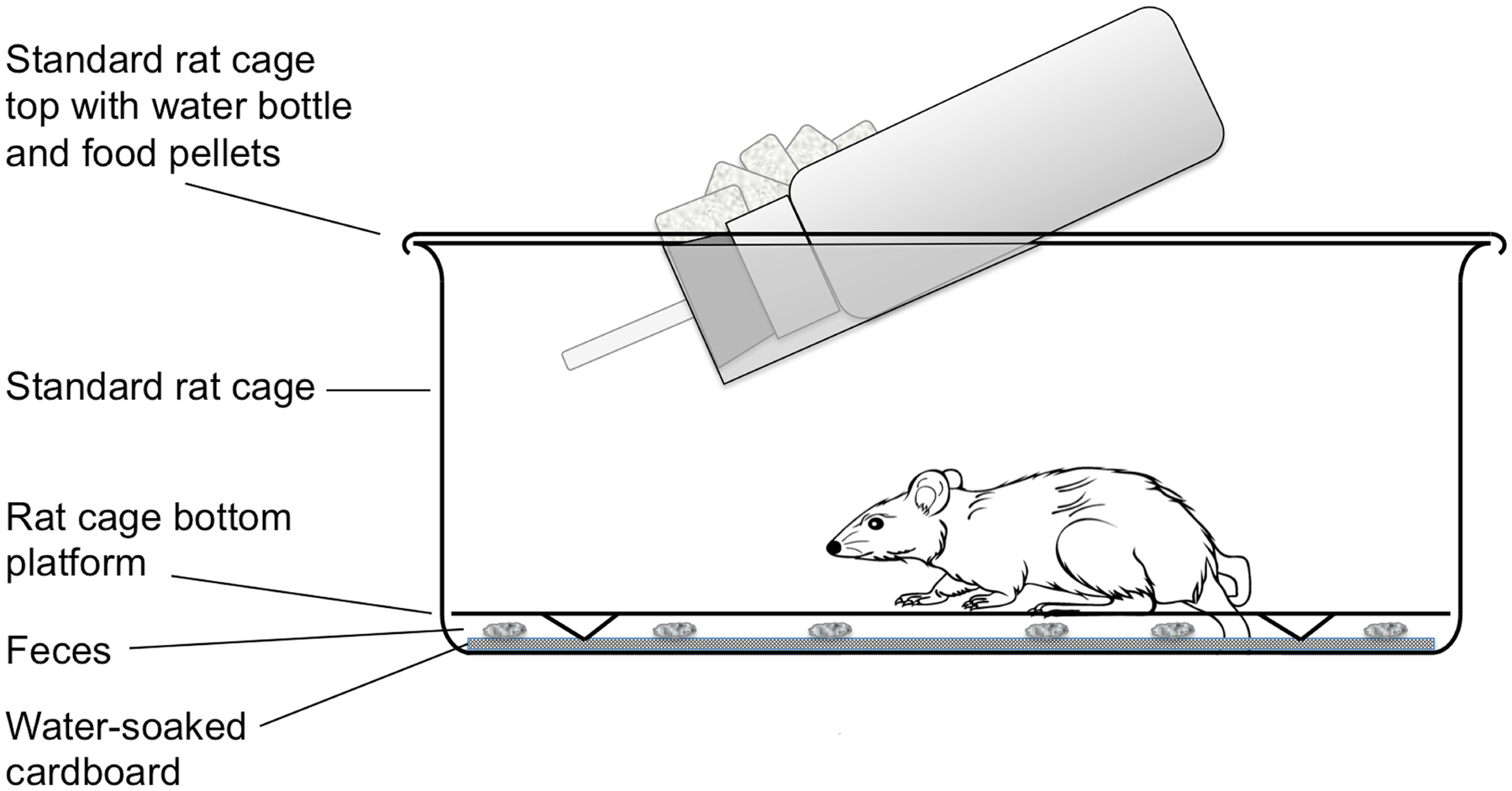
Methods for routine maintenance of S. stercoralis, for isolation of selected parasite stages en masse, for gene transfer by gonadal microinjection and for laser ablation of chemosensory neurons are detailed in another WormMethods article (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology). These methods are largely transferable to S. ratti, which, along with Brugia spp., is the subject of this article. The principal differences between rearing and maintenance of S. ratti and S. stercoralis are that the definitive host of S. ratti is the rat, whereas that of S. stercoralis is the dog or the human. Gerbils may act as an alternate laboratory host for both parasites. Secondly, collection of feces from gerbils and rats infected with S. ratti involves temporarily transferring these animals from their standard laboratory cages to cages designed for collection of fresh feces. This modified cage design is diagrammed in Figure 3.
|
Figure 3. Large rodent cage modified for overnight collection of feces from rats or gerbils infected with Strongyloides spp. The unit consists of a standard laboratory rat cage outfitted with water bottle and food pellets. For overnight fecal collection, standard bedding is replaced with a raised wire floor (Ancare, Bellmore, NY USA, product code N10SSRWF) underlain by a layer of cardboard saturated with deionized water. The latter maintains fecal pellets in a fully hydrated state during the overnight collection so that parasites do not succumb to desiccation.
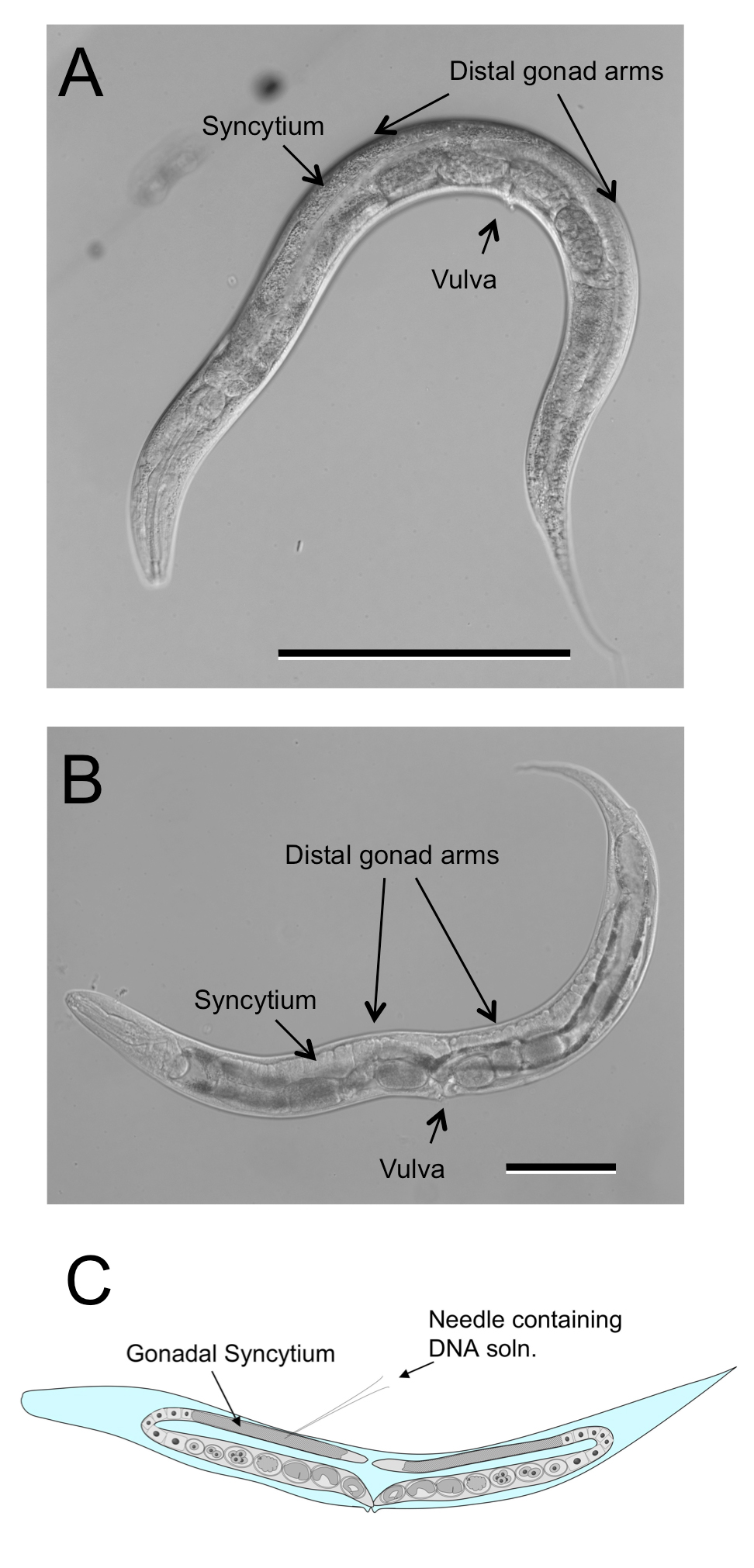
The free-living females of Strongyloides spp. and related genera are similar in their body plans (Figure 4B) and culture biologies to C. elegans hermaphrodites (Figure 4A). Their gonads, like those of C. elegans, are didelphic, taking the form of two symmetrical recurved tubes joining at midbody in the vulva. The germlines of both C. elegans hermaphrodites and Strongyloides free-living females are syncytial in the region just proximal to the distal tip cells and this common morphology has made it relatively straightforward to adapt standard methods for gonadal microinjection of DNA from C. elegans to Strongyloides spp (Figure 4C).
|
Figure 4. Free-living female of Strongyloides ratti compared to C. elegans hermaphrodite. A Hermaphrodite of C. elegans depicting double recurved gonad. B Free-living female of S. ratti depicting similar overall body size and architecture of gonad. C Diagrammatic representation of microinjection site common to both C. elegans hermaphrodites and Strongyloides free-living females. Scale bars in panels A and B = 100 μm.
Inverted microscope with gliding stage, 40X DIC objective and 10X bright field objective (e.g., Olympus Model IX70)
Micromanipulator (e.g., World Precision Instruments - WPI model MMJR)
Pressure regulator (e.g., WPI model PV820 Pneumatic Picopump)
Micropipet puller (e.g., Sutter Instrument Co. model P-30)
Stereo microscope with coaxial fluorescence (e.g., Olympus model SZX12)
Standard stereo microscopes (e.g., Olympus model SZX7)
Low temperature incubator set at 22 °C
Modified rat and gerbil cage (Figure 4)
Baermann apparatus (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology)
Compressed nitrogen
E. coli HB101
Standard supplies for C. elegans strain maintenance (see Maintenance of C. elegans)
60 mm NGM agar plates
E. coli OP50
Platinum worm picks made from 32-gauge (Kitco, #PT99WIRE0203) and 36-gauge (cat. no. PT99WIRE0127) platinum wire .
Microscope slides, unfrosted
50 mm coverslips
Halocarbon oil (Halocarbon Products Corp, River Edge, NJ USA, #9002083-9)
BU Saline
Na2HPO4 (50 mM)….0.71 g
KH2PO4 (22 mM)……0.30 g
NaCl (70mM)………..0.41 g
Water ………………..100 ml
Methyl prednisolone acetate
NGM Agar
2 % Agarose
Three days prior to microinjection, collect fresh feces from S. ratti-infected rats or gerbils by placing animals in modified rat cages overnight (Figure 3). Animal welfare regulations in the USA prohibit housing of rats or gerbils in the modified cages for periods longer than 24 hours.
Prepare charcoal coprocultures of infected host feces as described (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology). Incubate coprocultures at 22 °C.
On the day of microinjection, isolate free-living adults of S. ratti from fecal coprocultures by Baermann funnel (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology). Transfer the pellet of isolated free-living adults to a 60 mm NGM OP50 plate. Allow transfer medium to dry on the plate. It is important to use appropriately aged worms for injection. Select free-living females with 4-6 eggs per uterine arm to optimize transformation efficiency and worm survival.
Using published techniques (Mello and Fire, 1995; see also Transformation and microinjection and Strongyloides stercoralis: a model for translational research on parasitic nematode biology), microinject plasmid mix into one gonad arm of as many parental free-living females as possible. Plasmid mixes would typically contain donor and helper plasmids incorporating elements of the piggyBac transposon at 50 ng/µl and 25 ng/µl, respectively (note: rationale for transposon-mediated integrative transgenesis is discussed in Section 2.3.1). Experienced personnel in the Lok lab can prepare parasites for microinjection in the morning and inject approximately 40 parental female worms in the afternoon.
Pool all microinjected free-living females on a single NGM HB101 plate with a small particle of sterile rat feces; add free-living males at a proportion of approximately 1.5 males per female from the plate containing the Baermann funnel pellet. Incubate plate upside down in a humidified plastic box at 22 °C.
On days 5 to 6 following injection, examine the plate containing microinjected females using the fluorescence stereomicroscope. For plates containing transgene expressing L3i, harvest parasites by the Baermann funnel technique (see Transformation and microinjection and Strongyloides stercoralis: a model for translational research on parasitic nematode biology), substituting the plate contents for charcoal coproculture proceeds. This may be done by using a spatula to scoop the agar with parasites and bacterial lawn from the plate into the Kimwipe®-lined sieve of the Baermann funnel. Also, rinse both the plate and its lid into the Baermann funnel with a stream of warm (approx. 43 °C) deionized water. Once isolated, transgenic L3i may be pooled with the proceeds of one or two other microinjection sessions in a T-25 tissue culture flask containing 5 ml of BU saline and stored with flask horizontal at 25 °C prior to injection into susceptible rats.
Notes on modification of standard C. elegans microinjection technique
S. ratti is more subject to injury by microinjection than C. elegans, so personnel in the Lok lab find that the decreased number of transformed oocytes resulting from microinjecting only one gonadal arm is more than offset by the benefit of increased survivorship among microinjected worms.
S. ratti also appears somewhat more prone to fatal desiccation on dry agarose pads than C. elegans. It is common practice in the Lok lab to mount no more than two free-living S. ratti females at a time. By contrast, experienced C. elegans injectors can mount and inject several adult hermaphrodites at a time without risking fatal desiccation (Mello and Fire, 1995).
We have found that gently teasing microinjected S. ratti off the agarose microinjection pad with a 36 gauge platinum worm pick works as well as demounting the worms in a recovery buffer as described (Mello and Fire, 1995; see also Transformation and microinjection).
Initial attempts to establish stable lines of transgenic S. stercoralis and S. ratti using parental worms transformed with plasmid-based constructs alone failed, not for lack of germline transformation but rather because stably inherited transgenes were apparently silenced in the G2 generation of passage and beyond (Junio et al., 2008). We hypothesize that this apparent silencing is a result of plasmid-encoded transgenes being incorporated predominantly into episomal arrays as they are in C. elegans (Mello and Fire, 1995). As a remedy for this we have recently achieved chromosomal integration of transgenes in S. ratti using constructs incorporating elements of the piggyBac transposon system (Figure 5). This has allowed creation of the first stable transgenic lines of this parasite with sustained expression of transgenes in proportions of parasites approaching 100% (Shao et al. 2012 ). The following protocols for establishment of such lines assume that founding G1 transgenic larvae have transgenes that are integrated into the chromosomes by this method (See Protocol 1, Section 2.2.1.5).
|
Figure 5. Constructs for integration of transgenes into chromosomes of Strongyloides spp. A Plasmid pPV254 (GenBank accession # JX013636), example of a donor plasmid in which transgene coding sequence, in this case a gfp reporter driven by the Ss-act-2 promoter and terminated by the Ss-era-1 3′ UTR, is flanked by the inverted terminal repeats (ITR) of the piggyBac transposon. B Plasmid pPV402 (GenBank accession # JX013634), example of a helper plasmid encoding the piggyBac transposase expressed under the Ss-rps-21 promoter and terminated by the multifunctional Ss-era-1 3′ UTR. Parental free-living female Strongyloides are co-injected with donor and helper plasmid to effect chromosomal integration of the transgene construct.
Stereo microscope with coaxial fluorescence
Standard stereo microscopes
Modified rat and gerbil cage (Figure 4)
Baermann apparatus (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology)
Low temperature incubator set at 22 °C
Bone charcoal (5×8 grade, Ebonex, Melvindale, MI USA)
NGM agar plates
E. coli HB101
Autoclaved rat feces (2-3 g)
Glass depression slides (Erie Scientific Co., #1527)
Sterile 1 ml. syringes with 26-gauge s
Once at least 20 (preferrably 30-40) G1 (Table 1) transgenic S. ratti L3i (i.e., progeny of microinjected free living female worms) have been accumulated in BU saline at 25 °C (See protocol 1, Section 2.2.1.5, step 6), rinse a sterile 1 ml syringe with sterile BU saline; concentrate parasites into 100-300 μl volume and transfer to the barrel of the syringe. Attach a 26-gauge needle to the syringe and transport along with a tube containing 1 ml of sterile BU saline to the vivarium.
Inject transgenic L3i subcutaneously into an adult rat. Draw 0.5 ml of sterile BU into the same syringe and inject this subcutaneously into the rat to act as a rinse.
Draw the remaining 0.5 ml of sterile BU saline into the syringe and expel back into the eppendorf tube. Count any larvae in this aliquot of saline and subtract from the total number of parasites originally in the syringe to determine the actual number of L3i injected. Alternatively retained L3i may be injected into the same rat to maximize chances of infection with germline-transformed parasites.
Return the rat to the colony. S. ratti develops to patency (i.e., release of larval progeny in feces) in five days.
Beginning on day 5 after inoculation and then at 24 hour intervals thereafter, collect fresh feces from the inoculated rat using the modified rat cage as outlined in Protocol 1 (Section 2.2.1.5, step 1); establish charcoal coprocultures and incubate at 25 °C.
Incubate charcoal coprocultures for 24 hrs. at 25 °C and harvest G2 (Table 1) larvae (progeny of G1 worms undergoing the first passage in the rat) by the Baermann funnel technique (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology). Screen G2 larvae and free-living adults for transgene expression by fluorescence stereomicroscopy.
Transfer GFP-expressing G2 larvae and adults to NGM plates with HB101 lawns and small particles of sterile rat feces for mating and production of G3 progeny (Table 1). Incubate plates at 22 °C. (Note: Oxygenation of these cultures is important, so plates are covered but not sealed with parafilm as is frequently done for C. elegans cultures.)
At 7 days of incubation at 22 °C, screen NGM HB101 G3 larvae arising in plate cultures for transgene expression by fluorescence stereo microscopy. Pick all GFP expressing L3i and pool in depression slides containing sterile BU saline. Pool larvae collected each day in a T-25 flask of sterile BU saline as indicated in Protocol 1, Section 2.2.1.5, step 6, and store at 25 °C until at least 30 G3 transgenic L3i have been collected for inoculation.
Concentrate G3 L3i into a syringe and inoculate subcutaneously into a rat as indicated in steps 1-4 of this protocol.
Once the rat inoculated with G3 transgenic L3i is patent, collect coproculture larvae by the Baermann funnel technique as outlined in Steps 5-8 of this protocol to select G4 transgenic larvae from fecal culture (Table 1), establish mating pairs and rear G5 transgenic larvae on NGM HB101 plates with particles of rat feces.
In the G5 and beyond (Table 1), lines are further stabilized and maintained by repeated rounds of passage in rats and culture plates with selection on GFP. At the time of this submission we have maintained lines of transgenic S. ratti to the G10 generation with virtually 100% transmission and sustained expression of transgenes.
Table 1. Definition of generations* in derivation and maintenance of transgenic lines of Strongyloides ratti.
| GENERATION | DESCRIPTION |
|---|---|
| G0 | Free-living female microinjected with transgene construct. |
| G1 | Progeny of microinjected female, selected for transgene expression, cultured to infective third-stage larvae on plates, inoculated into susceptible host. |
| G2 | Progeny of G1 transgenic parasitic female in host (parthenogenetic); eliminated as first-stage larvae in host feces, selected for transgene expression and cultured to free-living males and females on plates. |
| G3 | Progeny of sexual cross between transgenic G2 free-living males and females, derived on plates, reared to infective larvae, selected for transgene expression and inoculated into susceptible host. |
| G4 | Progeny of G3 parasitic female in host (parthenogenetic), eliminated as first-stage larvae in host feces, cultured to free-living males and females on plates. |
| G5 and following | Alternating rounds of culture and host passage as in G2 – G4 above. Transgene expression frequency is usually 100% in G5 and following, obviating selection at each generation.** |
|
* Generations of passage are assigned “G” numbers as opposed to “F” or filial numbers since progeny of parasitic female worms arise by mitotic parthenogenesis and not through sexual crossing. ** Periodic monitoring and re-selection of lines for transgene expression is strongly recommended. |
|
Once stable lines of S. ratti have been prepared, they should be cryopreserved for long-term storage. We use a simple published method for this process (Nolan and Schad, 1992), which is similar in concept to standard methods for C. elegans (see Maintenance of C. elegans), but takes into account the greater susceptibility of S. ratti larvae to freezing damage relative to cultured C. elegans.
Baermann apparatus (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology)
Low temperature incubator set at 25 °C
Modified rat and gerbil cage (Figure 4)
−80 °C freezer
100 mm Petri dishes
100 mm filter paper circles (Whatman #5)
15 ml conical centrifuge tubes
2 ml cryovials
Activated charcoal
Wooden tongue depressors
Styrofoam shipping container
Collect fresh feces from infected rats or gerbils by placing in modified rat/gerbil cage (Figure 3). Feces are collected by scraping from the water soaked cardboard with a wooden tongue depressor.
Use freshly collected feces to prepare charcoal coprocultures in 100 mm Petri dishes as described (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology).
Incubate charcoal coprocultures at 25 °C for 7 days.
Isolate L3i from Day 5-7 coprocultures by the Baermann funnel technique (see Strongyloides stercoralis: a model for translational research on parasitic nematode biology). Transfer worm suspension to a 15 ml conical tube. Estimate concentration of parasites by counting worms in three 20 μl aliquots and averaging. Calculate total larvae isolated by the formula (No. parasites in 20 μl) × (50) × (total volume in ml). Generally we attempt to cryopreserve lots containing at least 10,000 L3i.
Wash L3i two times in deionized water; allow worms to gravity sediment between washes.
Suspend washed worm sediment in 1.0 ml of freezing solution containing 10% DMSO and 10% dextran in deionized water. Transfer worms in freezing solution to labeled 1.8 ml cryotube and allow to stand at room temperature for 1 hour.
Gradually cool cryotubes as described for C. elegans (see Maintenance of C. elegans) by transfering to a closed styrofoam shipping box and placing in the −80 °C freezer overnight. After gradual cooling, transfer cryotubes to permanent storage locations in the −80 °C freezer. As with C. elegans, it is recommended to thaw a test cryotube to assertain viability of the cryopreserved lot.
Thawing is as described (Nolan et al., 1988). Add 0.5 ml of cold (4 °C) RPMI to the cryotube and thaw in a 37 °C waterbath for approximately three minutes. Transfer contents of thawed cryotube to a 15 ml conical tube containing 10 ml of RPMI; spin at 150 x g for 7 minutes; discard supernatant and resuspend pellet in 3 ml. of RPMI for motility assessment.
Assess motility of thawed L3i at 20 hr post thawing and inoculate into rats (1000 L3i per rat) as described in protocol 2, Section 2.3.2.5, steps 1-4. We expect 10% recovery of normally motile larvae at 20 hr post thawing.
Brugia spp. are obligate parasites transmitted in nature by mosquito vectors in the genus Mansonia. Therefore, maintaining the full life cycle of B. malayi in the laboratory requires a colony of susceptible mosquitoes, usually a strain of Aedes aegypti selected for high susceptibility to Brugia infection, as well as susceptible vertebrate animal hosts, generally cats or gerbils. Although rearing Aedes aegypti and maintaining Brugia infection in laboratory animals are not technically difficult, they require secure, climate controlled insectary space, a source of whole blood (either a laboratory animal or other source) for blood feeding mosquitoes to support egg production, and at least one susceptible vertebrate animal species, most likely the gerbil, to maintain the parasite strain. While these capabilities would be necessary to perpetuate heritably transformed B. malayi produced in Protocol 5 below, transient transfection of B. malayi embryos can be accomplished with parasite material provided by the Filariasis Research Reagent Resource Center (FR3). Furthermore, transgenic adult B. malayi, which presumably express transgenes in promoter regulated fashion, as well as their transgenic microfilarial progeny could be produced by Protocol 4 below, using parasite material from FR3 and laboratory gerbils.
B. malayi can be transfected by microinjection, bombardment or soaking with DNA-calcium co-precipitates (Higazi et al., 2002; Shu et al., 2003; Xu et al., 2011). Microinjection can be used to transfect developing embryos in adult female parasites (Higazi et al., 2002). However, this process is technically difficult and injection irreversibly damages the cuticle of the parasite. Biolistic bombardment may be used to transfect infective larvae and adult females (Higazi et al., 2002). Biolositic transfection of intact parasites is also relatively inefficient, leading to poor expression of reporter genes. In contrast, transient transfection of isolated embryos from adult females is very efficient, leading to high levels of expression of reporter genes (Shu et al., 2003). Although the isolated embryos are developmentally incompetent, they may be maintained in culture for up to 10 days without a significant loss of viability (Shu et al., 2003). This technique has been employed in several studies to map the core promoter domains and regulatory region of various B. malayi promoters (de Oliveira et al., 2008; Higazi et al., 2005; Liu et al., 2009; Liu et al., 2007; Shu et al., 2003), as well as to study cis-acting signals necessary for trans splicing of B. malayi mRNAs (Higazi and Unnasch, 2004; Liu et al., 2010; Liu et al., 2007; Shu et al., 2003).
Mechanical methods of transfecting B. malayi have all been found to damage the parasite cuticle, resulting in parasites that are killed when implanted into the host. Thus, mechanical transfection methods cannot be used to develop transfected parasites that can develop throughout the subsequent life cycle stages. Recent studies have suggested that molting parasites are capable of taking up both exogenous RNA and DNA molecules (Song et al., 2010; Xu et al., 2011). This finding has been exploited to develop an “in-squito” method for inducing the uptake of interfering RNA (RNAi) by developing larvae in the mosquito vector of B. malayi (Song et al., 2010), and has been used to transfect B. malayi during the L3-L4 molt (Xu et al., 2011). The latter method, while inefficient, does result in the production of developmentally competent parasites which maintain transgenic sequences and reporter gene activity into the F1 generation (Xu et al., 2011).
Biolistics unit:
PDS-1000/He System BioRad laboratories #165-2257
Desiccant chamber:
Plastic box with a layer of CaCl2 covered with filter paper (Whatman® 3MM)
Humid chamber:
Plastic box containing paper towels soaked in distilled water
Vacuum Source:
A vacuum system capable of 100 l/min capacity. The system must be capable of maintaining a vacuum of at least 12.5 inHg.
Microcentrifuge
Tissue culture incubator
Vortex mixer fitted with a vertical tube holder
USA Scientific, #7404-9524
1100 PSI rupture discs BioRad #165-2329.
Stopping Screens BioRad #165-2336.
0.6 μm Gold Microcarriers BioRad #165-22623.
Macrocarriers BioRad #165-2335
Grade 4.5 to 5.0 helium (minimum 99.995% pure)
10 cm plastic petri dishes
3.5 cm plastic petri dishes
Sterile disposable curved blade scalpels
Sterile disposable transfer pipets
Adult female Brugia malayi: Five adult females are required per transfection. Parasites can be obtained through the Filariasis Research Reagent Resource Center (FR3). Contact the FR3 for availability.
All reagents are molecular biology grade, unless otherwise noted
70% (v/v) ethanol
Nuclease free water
50% (w/v) glycerol
2.4 M CaCl2
0.1 M spermidine. (made with free base spermidine, tissue culture grade from Sigma-Aldrich®, # S4139). Prepare this solution by adding 67.8 ml of sterile water to the contents of a vial containing 1 g of free base spermidine. Aliquot the solution into 0.5 ml aliquots in 1.5 ml microcentrifuge tubes and store them at −80 oC. Use each aliquot once and discard.
Absolute ethanol
Plasmid DNA solution (2 mg/ml in water). Plasmid DNA should be prepared using an endotoxin free purification method (such as the Qiagen Endo-free Maxi kit, #12362).
CF-RPMI medium:
Gibco RPMI 1640 medium with Hepes (Invitrogen™ #A10491-01) containing the following additives: 20% fetal calf serum, 20 mM glucose, 24 mM sodium bicarbonate, 2.5 μg/ml amphotericin B, 10 units/ml penicillin, 10 units/ml streptomycin and 40 μg/ml gentamicin.
Weigh out roughly 30 mg of gold beads (0.6 μm) into a 1.5 ml microfuge tube.
Add 1 ml of 70% ethanol (v/v) prepared with 100% HPLC grade ethanol and sterile deionized water.
Strap the tube on the platform vortex and mix for 15 minute.
Allow the particles to soak for 15 minutes at room temperature.
Pellet the beads in a microfuge for 5 seconds.
Discard the supernatant and resuspend the beads in 1 ml sterile nuclease free water.
Vortex at full speed for 1 minute.
Allow the particles to settle for 1 minute.
Pellet the beads by spinning for 3 sec. in the microfuge.
Discard the supernatant.
Repeat steps 7-9 two more times.
Resuspend the beads in 500 μl of 50% glycerol. This gives a concentration of roughly 60mg/ml. Store the beads at 4 oC.
Place the tube containing the prepared beads on to the platform vortex and mix for 15 minutes.
Remove 50 μl of the homogenized solution and transfer to a new 1.5 ml centrifuge tube.
Place the new tube with the top opened on the platform vortex. Add in order while continuing to vortex:
10 μl of 2 mg/ml purified DNA in water.
55 μl of 2.5 M CaCl2
22 μl of 0.1 M spermidine
After all of the ingredients are added, continue to mix the sample on the vortex for three minutes more.
Allow the beads to settle for 1 minute.
Spin out the beads for 2 seconds in the microcentrifuge (full speed).
Discard the supernatant. Add 280 μl of 70% ethanol. Rinse but do not resuspend the pelleted beads.
Decant off the liquid and add 280 μl of absolute ethanol.
Decant off the liquid and resuspend the beads in 18 μl of absolute ethanol.
Begin with five adult female worms per transfection. Collect the worms into a minimal amount of medium by decanting the medium into a dish. Remove as much of the medium as possible using a transfer pipet.
Dice the worms using a curved bladed scalpel to release the embryos.
Collect the embryos into a 1.5 ml microcentrifuge tube using CF-RPMI. Rinse the dish and add the rinse to the tube.
Spin the embryos at 6000 × g in the microcentrifuge for 3 minutes at room temperature to collect them.
Resuspend the embryos in 30 μl CF-RPMI.
Soak the 1100 PSI break disks in isopropanol and drain them off in a 10 cm petri dish with a macrocarrier holder in the center to prop the disks on their edges.
Load the macrocarrier with 6 μl bead suspension and place in into the dessicator box for five minutes.
Load the 1100 PSI break disk into the top of the unit and the macrocarrier into the macrocarrier holder.
Spread the embryos into a 1 cm circle in the bottom of a 3.5 cm dish. Place this dish into a 10 cm petri plate and place the dishes on to the platform located at the first position of the unit.
Set the gap distance of the disc retaining cap at 3/8 inch and the stopping screen support at the bottom of the launch assembly, providing a 16 mm macrocarrier travel distance.
Turn on the vacuum. Allow the vacuum in the chamber to reach 12.5 inches of mercury. Place the vacuum switch in the hold position.
Fire the unit, release the vacuum and remove the sample.
Place the dish containing the embryos into the humid box and cover. Let sit for five minutes.
Add 5 ml of CF-RPMI to the dish. Place the dish at 37 oC under 5% CO2 in the tissue culture incubator. Incubate the embryos for 48 hours without a media transfer to permit reporter gene expression.
Biosafety cabinet
Vortex mixer fitted with a vertical tube holder (USA Scientific, #7404-9524)
Sterile air supply
24-well tissue culture plates
Microcentrifuge tubes
3 ml sterile syringe fitted with a 20-gauge needle
One male gerbil 10 weeks of age per transfection. Note: male gerbils are more susceptible to infection by B. malayi than females (Ash, 1971).
250 Brugia malayi L3 per transfection. Parasites can be obtained through the Filariasis Research Reagent Resource Center (FR3). Contact the FR3 for availability.
All are molecular biology grade.
Minimum Essential Medium Alpha (Invitrogen #12571) with the following additives: 1000 units/mL of penicillin-streptomycin (Invitrogen) 10 μg/ml gentamycin (Sigma-Aldrich) 2 μg/ml ciprofloxin (Bayer) and 1 μg/mL ceftazidime (GlaxoSmithKline)
2 M CaCl2
Plasmid DNA solution (2 mg/ml). Plasmid DNA should be prepared using an endotoxin free purification method (such as the Qiagen Endo-free Maxi kit, #12362).
2× HBS: 50 mM HEPES (pH7.1) 280 mM NaCl 1.5 mM Na2HPO4. Final pH should be 7.1
RPMI 1640 medium with Hepes (Invitrogen #A10491-01)
Remove the L3 from the medium that they were shipped in by chilling them on ice for 5 minutes, decanting off the old medium from the settled L3, and replacing the medium with 600 μl of fresh RPMI 1640 medium.
Transfer the L3 and medium to a single well of a 24 well tissue culture plate.
For each transfection, prepare the two solutions in separate 1.5 ml microcentrifuge tubes:
DNA solution
plasmid DNA 3 μl
2 M CaCl2 18 μl
Water 29μl
Total volume 150 μl
2× HBS 150 μl
Working in a tissue culture hood, place the tube containing the 2× HBS in the platform of the Vortex mixer, and gently vortex it. The speed should be adjusted such that the tube can be vortexed safely with the cap off and can accommodate the addition of the prepared DNA solution. Continue to vortex while slowly adding the prepared DNA solution dropwise to the 2× HBS. Bubble air with a glass Pasteur pipet through the 2× HBS solution while slowly adding the CaCl2/DNA solution. When the DNA addition is complete, the solution should appear slightly opaque due to the formation of a final calcium phosphate-DNA co-precipitate.
Incubate the solution at room temperature for 30 minutes.
While swirling the plate containing the L3, add the DNA precipitate solution dropwise to the L3.
Load the solution containing the L3 and DNA precipitate into a 3 ml syringe with a 20-gauge needle. Slowly inject the mixture into the peritoneal cavity of a gerbil.
Maintain the gerbils under normal housing conditions for 120 days without disturbing them. Transgenic microfilaria may be recovered from the gerbils by peritoneal lavage employing 5 ml of PBS. Adult parasites may be recovered from the peritoneal cavity of euthanized animals.
Some future applications of transgenesis in S. stercoralis and other animal parasitic nematodes will involve experimental disruption or augmentation of gene expression or function. Examples of this would be in situ expression of interfering RNAs or of gene constructs designed to give dominant loss- or gain-of-function in target genes. Manipulations of this type could adversely affect development or viability of the parasites to the extent that it will be essential to express the transgenes involved conditionally. Presently there are no methods for regulatable transgene expression in parasitic nematodes. However, we are exploring an approach recently pioneered in malaria parasites (Muralidharan et al., 2011) in which transgene products are tagged with the degradation domain of E. coli dihydrofolate reductase which in turn is stablized by the folate analog antibiotic trimethoprim. In the absence of trimethoprim, recombinant proteins are targeted for degradation in the proteosome. This system is a particularly appealing for parasitology because trimethoprim may be administered to isolated parasites in vitro or safely in the drinking water of infected host animals.
The method for chemically mediated transfection presented above is more versatile than biolistics, as it permits the study of transgene expression in developmentally competent parasites, and can be used to study expression in all life cycle stages. However, this process is relatively inefficient. It is possible that changes in the procedure for chemically mediated transfection might result in an increase in its efficiency. For example, alternative chemical or biological methods may be more efficient than the calcium-phosphate - DNA coprecipitates reported here. Similarly, pantropic lentiviral or retroviral systems might also increase the efficiency of the transfection process. Currently it is not known in what form the transgenic sequences are maintained in B. malayi. However, in many other nematodes, including Caenorhabditis elegans, transgenic sequences are often maintained as large extrachromosomal arrays (Mello et al., 1991). These arrays, while heritable, are not inherited in a Mendelian fashion. Transgenic methods that result in stable integration of the transgenic sequences into the nuclear genome are therefore desirable for the production of stable parasite lines. Several techniques have been reported to encourage integration of transgenic sequences, including the use of heterologous transposons (Morales et al., 2007) pseudotyped retroviruses (Kines et al., 2008) and including homologous genomic DNA as a carrier during transfection (Schlager et al., 2009). Utilizing the in vivo transfection method described here in conjunction with one of these systems to develop an integrative transfection method is a logical next step in the effort to develop stable transgenic lines of B. malayi.
Ash, L.R. (1971). Preferential susceptibility of male jirds (Meriones unguiculatus) to infection with Brugia pahangi. J. Parasitol. 57, 777-780. Abstract
Ash, L.R., and Riley, J.M. (1970). Development of subperiodic Brugia malayi in the jird, Meriones unguiculatus, with notes on infections in other rodents. J. Parasitol. 56, 969-973. Abstract
Brooker, S. (2010). Estimating the global distribution and disease burden of intestinal nematode infections: adding up the numbers–a review. Int. J. Parasitol. 40, 1137-1144. Abstract Article
Charlier, J., Hoglund, J., von Samson-Himmelstjerna, G., Dorny, P., and Vercruysse, J. (2009). Gastrointestinal nematode infections in adult dairy cattle: impact on production, diagnosis and control. Vet. Parasitol. 164, 70-79. Abstract Article
de Oliveira, A.D., Katholi, C.R., and Unnasch, T.R. (2008). Characterization of the promoter of the Brugia malayi 12kDa small subunit ribosomal protein (RPS12) gene. Int. J. Parasitol. 38, 1111-1119. Abstract Article
Higazi, T.B., DeOliveira, A., Katholi, C.R., Shu, L., Barchue, J., Lisanby, M., and Unnasch, T.R. (2005). Identification of elements essential for transcription in Brugia malayi promoters. J. Mol. Biol. 353, 1-13. Abstract Article
Higazi, T.B., Merriweather, A., Shu, L., Davis, R., and Unnasch, T.R. (2002). Brugia malayi: Transient transfection by microinjection and particle bombardment. Exp. Parasitol. 100, 95-102. Abstract Article
Higazi, T.B., and Unnasch, T.R. (2004). Intron encoded sequences necessary for trans splicing in transiently transfected Brugia malayi. Mol. Biochem. Parasitol. 137, 181-184. Abstract Article
Hotez, P.J., Fenwick, A., Savioli, L., and Molyneux, D.H. (2009). Rescuing the bottom billion through control of neglected tropical diseases. Lancet. 373, 1570-1575. Abstract Article
Junio, A.B., Li, X., Massey, H.C., Jr., Nolan, T.J., Todd Lamitina, S., Sundaram, M.V., and Lok, J.B. (2008). Strongyloides stercoralis: cell- and tissue-specific transgene expression and co-transformation with vector constructs incorporating a common multifunctional 3’ UTR. Exp. Parasitol. 118, 253-265. Abstract Article
Kines, K.J., Morales, M.E., Mann, V.H., Gobert, G.N., and Brindley, P.J. (2008). Integration of reporter transgenes into Schistosoma mansoni chromosomes mediated by pseudotyped murine leukemia virus. FASEB J. 22, 2936-2948. Abstract Article
Krecek, R.C., and Waller, P.J. (2006). Towards the implementation of the “basket of options” approach to helminth parasite control of livestock: emphasis on the tropics/subtropics. Vet. Parasitol. 139, 270-282. Abstract Article
Liu, C., Chauhan, C., Katholi, C.R., and Unnasch, T.R. (2009). The splice leader addition domain represents an essential conserved motif for heterologous gene expression in B. malayi. Mol. Biochem. Parasitol. 166, 15-21. Abstract Article
Liu, C., Chauhan, C., and Unnasch, T.R. (2010). The role of local secondary structure in the function of the trans splicing motif of Brugia malayi. Mol. Biochem. Parasitol. 169, 115-119. Abstract Article
Liu, C., de Oliveira, A., Higazi, T.B., Ghedin, E., DePasse, J., and Unnasch, T.R. (2007). Sequences necessary for trans-splicing in transiently transfected Brugia malayi. Mol. Biochem. Parasitol. 156, 62-73. Abstract Article
Liu, C., Oliveira, A., Chauhan, C., Ghedin, E., and Unnasch, T.R. (2010). Functional analysis of putative operons in Brugia malayi. Int. J. Parasitol. 40, 63-71. Abstract Article
Mello, C., and Fire, A. (1995). DNA transformation. Methods Cell Biol. 48, 451-482. Abstract
Mello, C.C., Kramer, J.M., Stinchcomb, D., and Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. Abstract
Morales, M.E., Mann, V.H., Kines, K.J., Gobert, G.N., Fraser, M.J. Jr., Kalinna, B.H., Correnti, J.M., Pearce, E.J., and Brindley, P.J. (2007). piggyBac transposon mediated transgenesis of the human blood fluke, Schistosoma mansoni. FASEB J. 21, 3479-3489. Abstract Article
Muralidharan, V., Oksman, A., Iwamoto, M., Wandless, T.J., and Goldberg, D.E. (2011). Asparagine repeat function in a Plasmodium falciparum protein assessed via a regulatable fluorescent affinity tag. Proc. Natl. Acad. Sci. U. S. A. 108, 4411-4416. Abstract Article
Nolan, T.J., Aikens, L.M., and Schad, G.A. (1988). Cryopreservation of first-stage and infective third-stage larvae of Strongyloides stercoralis. J. Parasitol. 74, 387-391. Abstract
Nolan, T.J., and Schad, G.A. (1992). Cryopreservation of infective third-stage larvae of Strongyloides ratti. J. Helm. Soc. Wash. 59, 133-135.
Schlager, B., Wang, X., Braach, G., and Sommer, R.J. (2009). Molecular cloning of a dominant roller mutant and establishment of DNA-mediated transformation in the nematode Pristionchus pacificus. Genesis 47, 300-304. Abstract Article
Shao, H., Li, X., Nolan, T.J., Massey, H.C. Jr., Pearce, E.J., and Lok, J.B. (2012). Transposon-mediated chromosomal integration of transgenes in the parasitic nematode Strongyloides ratti and establishment of stable transgenic lines. PLoS Pathog. 8, e1002871. Abstract Article
Shu, L., Katholi, C., Higazi, T., and Unnasch, T.R. (2003). Analysis of the Brugia malayi HSP70 promoter using a homologous transient transfection system. Mol. Biochem. Parasitol. 128, 67-75. Abstract Article
Song, C., Gallup, J.M., Day, T.A., Bartholomay, L.C., and Kimber, M.J. (2010). Development of an in vivo RNAi protocol to investigate gene function in the filarial nematode, Brugia malayi. PLoS Pathog. 6, e1001239. Abstract Article
*Edited by Ralf Sommer. Last revised February 28, 2013, Published September 30, 2013. This chapter should be cited as: Lok J. B. and Unnasch T. R. Transgenesis in animal parasitic nematodes: Strongyloides spp. and Brugia spp. (September 30, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.163.1, http://www.wormbook.org.
Copyright: © 2013 James B. Lok and Thomas R. Unnasch. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. Email: jlok@vet.upenn.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.