Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Receptors of the Notch family mediate cell-cell interactions during animal development, and aberrations in Notch signaling have been implicated in human disease. Studies in Caenorhabdits elegans have made essential contributions towards understanding the biological roles and molecular mechanism of this fundamental signaling system. A major development in the field since the original version of this chapter (LIN-12/Notch signaling in C. elegans) has been an explosion in information about the structural biology of Notch signaling; crystallographic determinations of structures, including structures of C. elegans components, have contributed much to the current understanding of molecular mechanism. Thus, here, we not only cover the genetics of Notch in C. elegans, focusing on conserved core components and modulators, we have also included structural information about these components, describing the key events occurring during ligand binding and transcriptional control of target genes. In addition to text, we include Tables listing core components and key modulators of the signaling pathway along with their orthologs in Drosophila and mammals, a Table listing validated target genes in various processes in C. elegans, and animated features to illustrate structural attributes.
Notch is a fundamental signaling system that mediates cell-cell interactions in animal development, and aberrations in Notch signaling or components of the signaling system underlie human disease. Genetic analysis of Notch signaling in C. elegans has illuminated universal aspects of this essential conserved pathway. Although the two C. elegans Notch genes, lin-12 and glp-1, are best known for their roles in cell fate decisions during development, recent studies have expanded the roles of these genes in C. elegans beyond cell fate to other types of events including the regulation of tubular morphogenesis (Rasmussen et al., 2008), dauer maintenance and recovery (Ouellet et al., 2008), the response to anoxia (LaRue and Padilla, 2011), behavior (Singh et al., 2011), oocyte growth (Nadarajan et al., 2009) and cell fate reprogramming (Jarriault et al., 2008). Thus, ongoing investigations into Notch signaling in C. elegans have implications for many different aspects of development and physiology.
This chapter contains five additional sections. In Section 2, we briefly summarize the key events during signal transduction by Notch. In Section 3, we describe the conserved core components of the Notch signaling system and their genetic identification in C. elegans. In Section 4, we consider detailed aspects of the mechanism illuminated by structural studies of the C. elegans and mammalian core components individually and in complexes. In Section 5, we list target genes in C. elegans and comment on a potential difference in binding site architecture between C. elegans and other animals. In Section 6, we summarize genetic screens for modulators of Notch signaling in C. elegans.
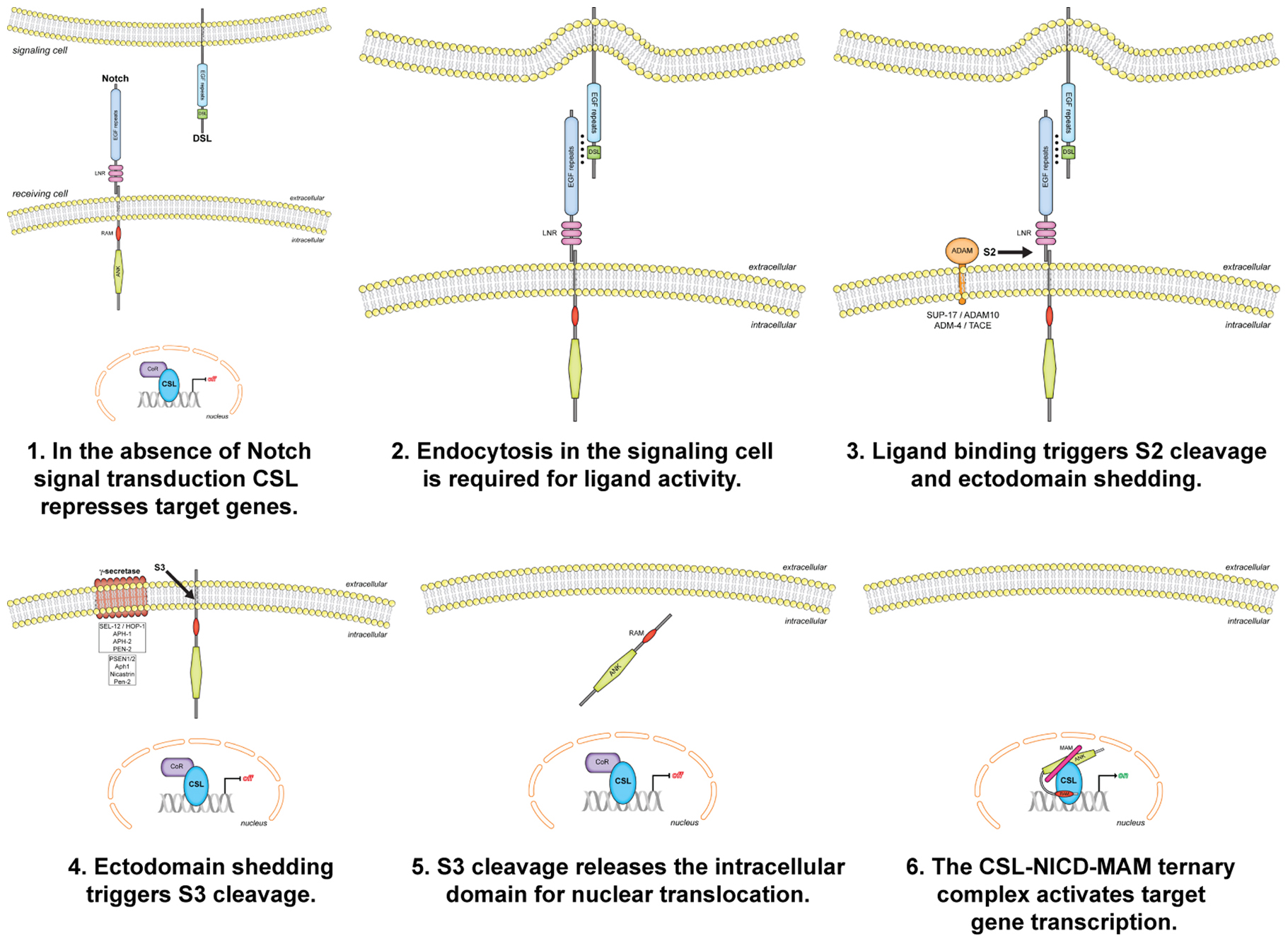
All Notch proteins have the arrangement of amino acid sequence motifs illustrated in Figure 1. The intracellular domain of Notch is essentially a membrane-tethered transcription factor whose release is regulated by ligand binding. Release of the intracellular domain involves two proteolytic cleavage steps (Figure 1). The first cleavage is regulated: binding of a ligand of the DSL family exposes a cleavage site (“site 2”) in the ectodomain, making it accessible to an ADAM family protease. The second cleavage is constitutive: the remaining transmembrane protein, with only a short extracellular stub, is a substrate for γ-secretase, which cleaves within the transmembrane domain at “site 3”. The intracellular domain, released from its transmembrane tether, translocates to the nucleus. In the nucleus, the intracellular domain forms a complex with a sequence-specific DNA binding protein of the CSL family and a co-activator of the Mastermind family to promote target gene expression.
This basic model was first proposed after observing that expression of the intracellular domains of LIN-12 in C. elegans and Notch in Drosophila caused phenotypes associated with constitutive activation of the receptors (Struhl et al., 1993). For a detailed discussion of how the cleavage model was supported, challenged, and finally accepted, as well as further information about the central role C. elegans played in understanding the role of Notch signaling in lateral specification (lateral inhibition), please see (Greenwald, 2012).
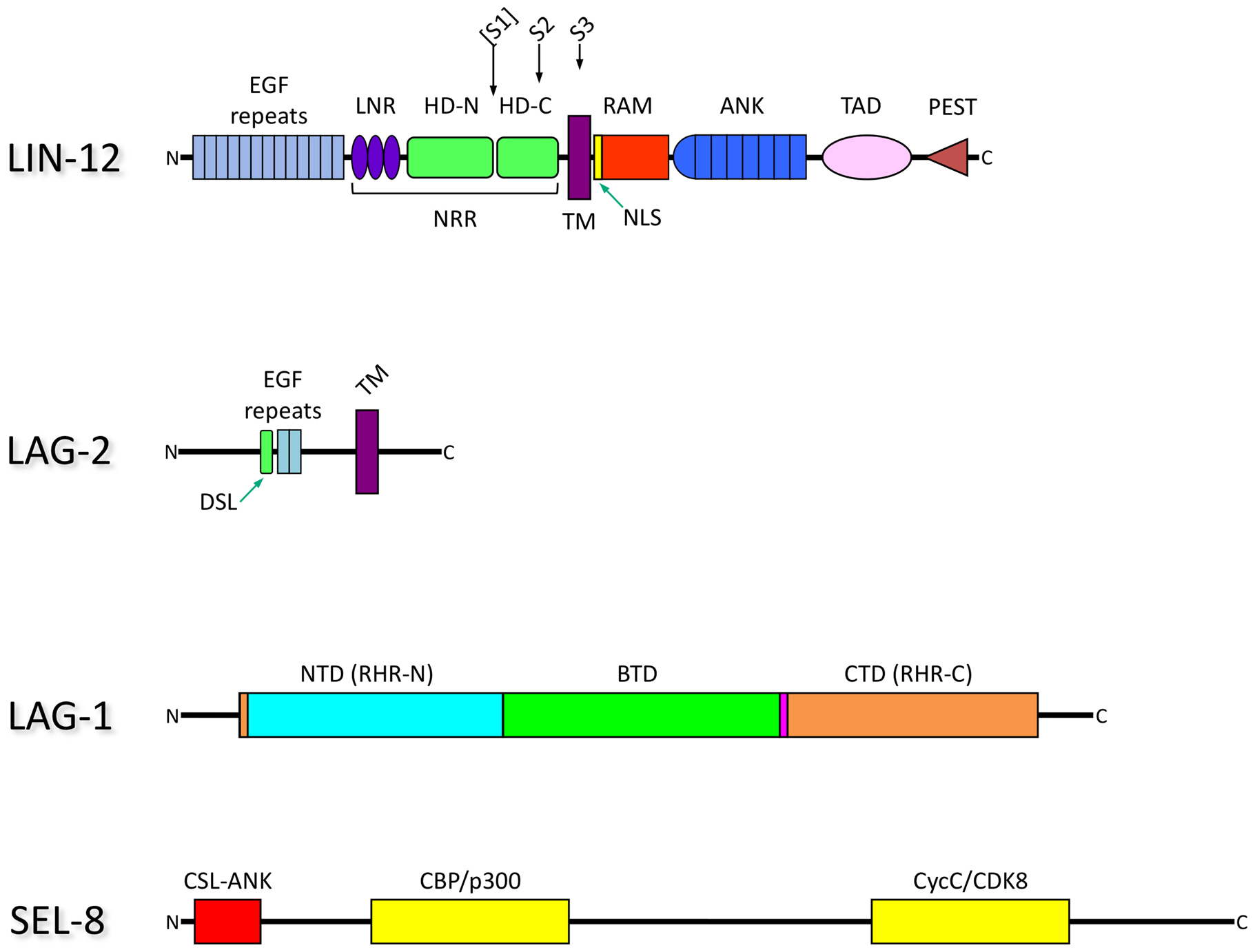
All of the core components of the Notch signaling system were identified through genetic analysis in C. elegans and Drosophila. Genetic studies in C. elegans first linked γ-secretase activity to Notch signaling and defined all four γ-secretase core components. Ligands and nuclear complex components were first identified in Drosophila, but genetic and structural studies of the C. elegans components led to significant insights into their functions (see below Section 4). The domain organization of LIN-12/Notch, the canonical ligand LAG-2, and the nuclear complex components is shown in Figure 2. Core components are also listed in Table 1, along with their Drosophila and mammalian orthologs.
|
Figure 2. Domain organization of core components from C. elegans.
LIN-12: The ectodomain is followed by a single transmembrane (TM) region and the intracellular domain (NICD). The S2 (ADAM protease) and S3 (γ-secretase) cleavage sites are indicated. The potential, but not formally demonstrated, S1 cleavage site in LIN-12 is shown in parentheses. GLP-1 has a similar domain organization.
EGF, Epidermal Growth Factor (EGF)-like motif.
LNR, LIN-12/Notch Repeat.
HD, Heterodimerization Domain; shown as if Furin cleavage occurs at site 1, resulting in HD-N, amino terminal segment, HD-C, carboxy terminal segment. Cleavage at NRR, Negative Regulator Region. The NRR encompasses the LNR repeats and the HD domain.
TM, TransMembrane domain.
NLS, Nuclear Localization Sequence.
RAM, a region that mediates interactions with LAG-1.
ANK, Ankyrin repeat motifs.
TAD, Transcriptional Activation Domain.
LAG-2: The DSL ligand LAG-2 is composed of an ectodomain, a single transmembrane domain, and a short intracellular domain.
DSL, Delta Serrate LAG-2 domain.
EGF, EGF-like motif.
LAG-1: The CSL DNA-binding transcription factor consists of three domains.
NTD, Amino Terminal Domain.
CTD, Carboxy Terminal Domain. The NTD and CTD are often referred to as Rel Homology Region (RHR) -N and -C domains, due to their structural similarity to Rel proteins, such as NF-kappaB.
BTD. β-Trefoil Domain.
SEL-8: This component appears to be the counterpart of Drosophila or mammalian Mastermind, but shares little primary sequence similarity with them. Domains other than CSL-ANK are inferred based on Mastermind.
CSL-ANK, N-terminal domain that interacts with CSL and the ANK domain of NICD (CSL-ANK).
CBP/p300, domain that recruits the ubiquitous coactivator CBP/p300.
CycC/CDK8, domain that recruits the kinase CycC/CDK8.
Table 1. Core components of the Notch signaling system
| Gene | allele type | Notch phenotype | Suppress/enhance§ | Mammalian | C. elegans references |
|---|---|---|---|---|---|
| Receptors | |||||
| lin-12 | hypermorph* | 0 AC-Egl, Muv, other* | Notch1-4 | (Greenwald et al., 1983; Ferguson and Horvitz, 1985) | |
| null | 2 AC, Evl, other** | (Greenwald et al., 1983) | |||
| glp-1 | null (zygotic) | Glp*** | Notch1-4 | (Austin and Kimble, 1987) | |
| loss (maternal) | Mel (Aph or Apx)*** | (Priess et al., 1987; Mello et al., 1994) | |||
| hypermorph | Tum, Pro | (Berry et al., 1997; Pepper et al., 2003) | |||
| Ligands§§ | |||||
| lag-2 (sel-3) | null | Lag**** | (DSL) | (Lambie and Kimble, 1991) | |
| neomorph | Sup lin-12(d) | (Tax and Thomas, 1994; Tax et al., 1997) | |||
| apx-1 | loss | Mel (Apx) | (DSL) | (Mango et al., 1994; Mello et al., 1994) | |
| arg-1 | (DSL) | (Mickey et al., 1996) | |||
| dsl-1 | null | (DSL) | (Chen and Greenwald, 2004) | ||
| Ligand activity | |||||
| epn-1 | RNAi | Glp | Epsin | (Tian et al., 2004) | |
| Site 2 protease | |||||
| sup-17 | loss | Mel | Sup lin-12(d) | ADAM10 | (Tax et al., 1997; Wen et al., 1997) |
| adm-4 | ADAM17/TACE | (Jarriault and Greenwald, 2005) | |||
| Site 3 protease | |||||
| sel-12 | null | Egl | Sup lin-12(d) | Presenilin 1, 2 | (Levitan and Greenwald, 1995; Cinar et al., 2001) |
| hop-1 | null | none | Presenilin 1, 2 | (Li and Greenwald, 1997; Westlund et al., 1999) | |
| aph-1 | loss | Mel (Aph), Egl | Pen***** | Aph1 | (Francis et al., 2002; Goutte et al., 2002) |
| aph-2 | loss | Mel (Aph), Egl | Nicastrin | (Goutte et al., 2000; Levitan et al., 2001) | |
| pen-2 | loss | Pen | Pen2 | (Francis et al., 2002) | |
| Nuclear complex | |||||
| lag-1 | null | Lag | Sup lin-12(d) | CBF1/RBP-J | (Lambie and Kimble, 1991) |
| sel-8 (lag-3) | rare | Mel | Sup lin-12(d) | Mam | (Tax et al., 1997; Doyle et al., 2000) |
| RNAi | Lag | (Petcherski and Kimble, 2000) | |||
|
Alternate, unofficial gene names are given in parentheses. §identified in a suppressor or enhancer screen. §§Ligands are members of the DSL family; a similar domain structure is seen in Drosophila and mammalian ligands, but orthology is difficult to assign. *lin-12(d) alleles, see text. 0 AC-Egl, lack of an anchor cell prevents vulval induction. Muv, multivulva due to ectopic induction of the 2o vulval fate. For additional phenotypes, see Greenwald et al. (1983) and Wormbase. **lin-12(0) mutants have 2 anchor cells (2AC) and abnormal vulval eversion (Evl); for other phenotypes, see Greenwald et al. (1983) and Wormbase. ***Glp, germline proliferation defective; Mel, maternal effect lethal; Aph, anterior pharynx defective; Apx, anterior pharynx excess. See listed references and Wormbase for more details. ****larval arrest with cell fate transformations seen in the lin-12 glp-1 double mutant; see Lambie and Kimble (1991) and Wormbase for details. *****Pen, presenilin enhancer (Francis et al., 2002) |
|||||
LIN-12 and GLP-1 both have the hallmark arrangement of amino acid sequence motifs present in all Notch proteins (Yochem et al., 1988; Yochem and Greenwald, 1989) (Figure 1).
lin-12 was identified in a genetic screen for mutations that have defective vulval development (Greenwald et al., 1983; Ferguson and Horvitz, 1985). The first alleles isolated, called lin-12(d) alleles, were dominant and hypermorphic. Intragenic reversion of these dominant mutations yielded null alleles, called lin-12(0) (Greenwald et al., 1983). Phenotypic analysis indicated that lin-12(d) and lin-12(0) alleles cause opposite cell fate transformations in many different cell fate decisions, establishing the binary nature of Notch-mediated cell fate decisions (Greenwald et al., 1983). Further analysis suggested that lin-12(d) mutations result in ligand-independent constitutive activation and affect a negative regulatory domain or higher order structure (Greenwald and Seydoux, 1990); now, with structural information, the region affected by these mutations has been termed the “negative regulatory region” (NRR) (see Section 4). These lin-12(d) mutations are the prototype for a class of mutations that have been found to cause T- cell acute lymphoblastic leukemia (T-ALL) (Weng et al., 2004).
glp-1 was identified in two different, contemporaneous genetic screens. A screen for sterile mutants revealed that loss of “zygotic” glp-1 activity limits germline proliferation and causes germ cells to enter meiosis prematurely (Austin and Kimble, 1987; see also WormBook chapters Germline proliferation and its control and Notch signaling in the C. elegans embryo). Other screens based on germline aspects of glp-1 function yielded dominant alleles altering the NRR and causing continued mitotic proliferation of the germline at the expense of gamete production (Berry et al., 1997; Pepper et al., 2003). An allele that deletes the PEST domain also has increased activity (Mango et al., 1991) and resembles another class of T-ALL mutations (Weng et al., 2004).
A screen for maternal-effect embryonic lethal mutations revealed that loss of maternal glp-1 activity prevents cell fate induction at the 12-cell stage of embryogenesis (Priess et al., 1987); subsequently, several additional embryonic cell-cell interactions have been shown to require glp-1 or lin-12 activity (Priess, 2005).
The existence of two Notch genes in C. elegans allowed for a test of whether different Notch proteins are functionally redundant, which is of additional interest because vertebrates have four Notch proteins (and Drosophila has only one). Lambie and Kimble (Lambie and Kimble, 1991) showed that double mutants lacking zygotic lin-12 and glp-1 activity die as L1 larvae with various discrete defects, defining the “Lag” phenotype, for “Lin-12 and Glp-1.” Their results suggested that the two Notch proteins, though divergent in sequence, are functionally redundant (Lambie and Kimble, 1991). GLP-1 is able to substitute for LIN-12 in cell fate decisions, providing further evidence for true functional redundancy (Fitzgerald et al., 1993).
The Lag phenotype of the lin-12 glp-1 double mutant served as the basis of a genetic screen that identified two additional core components, lag-1 and lag-2 (Lambie and Kimble, 1991) (Table 1). These two genes, along with other core components and modulators, also emerged in genetic screens for extragenic suppressors and enhancers (Section 6).
“DSL” is an acronym derived from three canonical ligands for Notch: Drosophila Delta and Serrate, and C. elegans LAG-2. DSL proteins all contain one or more EGF motifs, as well as an amino terminal motif that is unique to the family, the “DSL motif” (Fig. 2).
The first C. elegans ligand identified, lag-2, was defined genetically in two ways: by null alleles that confer the Lag phenotype (Lambie and Kimble, 1991) and by neomorphic alleles that act as suppressors of lin-12(d) mutations (Tax and Thomas, 1994; Tax et al., 1997). The apx-1 (Mango et al., 1994; Mello et al., 1994), arg-1(Mello et al., 1994; Kostas and Fire, 2002) and dsl-1 (Chen and Greenwald, 2004; Hoyos et al., 2011) genes also encode ligands that have been genetically linked to discrete Notch-mediated events in C. elegans development.
C. elegans DSL proteins have two interesting differences as compared to ligands in other organisms. First, it is possible to engineer secreted forms that can activate LIN-12 or GLP-1 (Fitzgerald and Greenwald, 1995), forms that in other organisms generally act as dominant-negatives. In addition, there appear to be naturally secreted forms (Chen and Greenwald, 2004). However, it is not known whether these secreted forms function in association with membranes or require endocytosis for ligand activity (see below). Second, sequence differences at the amino termini of C. elegans ligands include the lack of a pair of variant EGF-like repeats, termed the “Delta and OSM-11” (DOS) motif, which is present in other DSL proteins (Komatsu et al., 2008) (see also Kovall and Blacklow, 2010). Instead, C. elegans contains genes that encode “DOS-only” proteins; the canonical member of the DOS-only family, OSM-11, appears to facilitate ligand activity in Vulval Precursor Cells (VPCs) and has been proposed to act as a co-ligand in this context (Komatsu et al., 2008). The generality of DOS-only protein function remains to be determined.
Extensive analysis in Drosophila and mammals has established Epsin as a core component required for ligand activity (e.g., Wang and Struhl, 2004). The role of Epsin-mediated endocytosis is an active area of research in the Notch field. RNAi experiments suggest that EPN-1, C. elegans Epsin, influences ligand activity (Tian et al., 2004), but an absolute requirement for Epsin in C. elegans (or for endocytosis per se) has not yet been assessed.
The intracellular domain of Notch, a CSL protein, and Mastermind form a transcriptional activation complex that promotes expression of target genes. Numerous other proteins, which play more general roles in transcriptional activation, are also recruited through interactions with this core complex.
CSL, an acronym for “CBF1/Su(H)/LAG-1”, is the sequence-specific DNA binding protein with which the intracellular domain of Notch proteins interact to promote target gene expression. Mammalian CBF1 (also called RBP-J) had been independently identified numerous times as a DNA binding protein in association with different promoters; the key connection of this protein to Notch was made through genetic studies of Drosophila Suppressor of Hairless [Su(H)]. In the absence of Notch activation, CSL functions as a transcriptional repressor, in a complex with co-repressors.
In C. elegans, lag-1 was defined by null mutations that cause a Lag phenotype, suggesting that lag-1 is an essential component for Notch signaling (Lambie and Kimble, 1991). Weaker alleles of lag-1 were subsequently also recovered in modifier screens (Qiao et al., 1995; Katic et al., 2005). Molecular cloning revealed that LAG-1 is a CSL protein and further analysis substantiated that it has the same binding site specificity as its Drosophila and mammalian orthologs (Christensen et al., 1996) (see below).
sel-8 was initially defined by neomorphic mutations that suppress lin-12(d) (Tax et al., 1997) (Doyle et al., 2000) and the SEL-8 protein (under the provisional name “LAG-3”) was also independently identified through a yeast two-hybrid screen for proteins that interact in a ternary complex with LAG-1 and the GLP-1 intracellular domain (Petcherski and Kimble, 2000). These interactions suggested that SEL-8 serves the same function as Drosophila and mammalian Mastermind (Petcherski and Kimble, 2000). Mastermind is an integral part of the transcriptional activation complex (Wu et al., 2000; Kitagawa et al., 2001) and may enhance phosphorylation and turnover of the Notch intracellular domain (Fryer et al., 2004).
Three proteolytic processing events have been defined for mammalian Notch. Proteolysis at site 1 by Furin during its trafficking to the surface is constitutive (Blaumueller et al., 1997; Logeat et al., 1998). It is not known whether LIN-12 and GLP-1 are processed at site 1, but Western blot analysis of GLP-1 is consistent with the possibility that it is (Crittenden et al., 1994). Drosophila Notch does not undergo this cleavage (Kidd and Lieber, 2002), suggesting that it is not pertinent to signal transduction per se.
Proteolysis of Notch at sites 2 and 3 leads to release of the intracellular domain for signal transduction. Site 2 is located in the ectodomain and is cleaved in a ligand-dependent manner. The site 2 protease is a member of the ADAM (A Disintegrin And Metalloprotease domain) protease family. In C. elegans, genetic analysis suggests that the main site 2 protease is encoded by sup-17. This gene was defined by hypomorphic alleles that suppress lin-12(d) mutations (Tax et al., 1997); molecular cloning and sequence analysis (Wen et al., 1997) established it as an ADAM protease orthologous to Drosophila Kuzbanian, also implicated in Notch signaling (Rooke et al., 1996; Pan and Rubin, 1997). The mammalian ortholog of SUP-17 and Kuzbanian is called ADAM10. Biochemical studies originally implicated a different mammalian ADAM protein, called TACE or ADAM17, in S2 cleavage of Notch (Brou et al., 2000). However, further studies in C. elegans and mammals suggest that both SUP-17/ADAM10 and ADM-4/TACE may function as S2 proteases and that there may be some functional redundancy between them (Jarriault and Greenwald, 2005).
Site 3 is located within the transmembrane domain. The site 3 protease, γ-secretase, can cleave within the transmembrane domain of virtually any type 1 membrane protein substrate with a short ectodomain (Struhl and Adachi, 2000). Thus, Notch becomes a substrate for γ-secretase as a result of the ligand-induced S2 cleavage. γ-secretase is a protein complex that consists of Presenilin, the catalytic subunit, and three other proteins that contribute to substrate recognition and stability: APH-2/Nicastrin, APH-1, and PEN-2. Expression of these four components together in yeast are sufficient to reconstitute γ-secretase activity (Edbauer et al., 2003).
Genetic analysis in C. elegans was critical for elucidating the components of the complex and the requirement of γ-secretase for Notch activity. Presenilin was initially connected to Notch when null alleles of sel-12 were identified as suppressors of lin-12(d) (Levitan and Greenwald, 1995). These null alleles are viable because of functional redundancy with a second Presenilin-encoding gene, hop-1. The essential requirement for Presenilin in Notch signaling was revealed by showing that concomitant loss of both sel-12 and hop-1 cause hallmark Lin-12, Glp-1 and Lag phenotypes, demonstrating that Notch-mediated signaling events throughout development depend on Presenilin activity (Li and Greenwald, 1997; Westlund et al., 1999).
In vivo topological studies in C. elegans defined the seventh transmembrane domain (Li and Greenwald, 1996; Li and Greenwald, 1998), which had not been identified by traditional topological studies or sequence analysis, and included what proved to be a key catalytic aspartyl residue. Definitive evidence that Presenilin has a catalytic function came from subsequent aspartyl protease inhibitor studies in mammalian cells.
The three other core components of γ-secretase were also identified genetically in C. elegans. APH-1 and APH-2 were identified by maternal-effect lethal mutations that confer the Anterior PHarynx defect resulting from failure of glp-1 signaling at the 12 cell stage of embryogenesis (Priess, 2005; Goutte et al., 2000; Goutte et al., 2002) (Table 1). Homozygous mutants are viable when maternally rescued. The mammalian ortholog of APH-2, Nicastrin, was independently identified through its physical association with Presenilin and initially assessed functionally in C. elegans by RNAi, revealing a role in Notch signaling (Yu et al., 2000). aph-1 as well as the fourth component of γ-secretase, pen-2, were identified in a “Presenilin enhancer” (Pen) screen for Notch loss-of-function phenotypes in a sel-12 mutant background (Francis et al., 2002) (Table 1). Biochemical analysis, principally in mammalian cell culture, has suggested that Nicastrin and Aph-1 form an initial complex, which binds to and stabilizes the Presenilin holoprotein; PEN-2 then associates with this tripartite complex to confer γ-secretase activity (reviewed in Iwatsubo, 2004). Endoproteolysis in the cytosolic loop of SEL-12/Presenilin (Li and Greenwald, 1996; Thinakaran et al., 1996; Li and Greenwald, 1998) is facilitated by PEN-2 and is believed to be important for γ-secretase activity (reviewed in Iwatsubo, 2004), although deletion of this region does not appreciably abrogate Presenilin activity in C. elegans (Levitan et al., 1996).
Following the identification of the core signaling components from genetic studies in C. elegans and Drosophila, there has been much interest in determining high-resolution structures of these components, and the complexes they form, in order to provide further molecular insights into the mechanism of signal transduction. In particular, there have been three areas in which significant progress has been made: (1) the structures of the Notch and DSL EGF repeats that are involved in receptor-ligand complexation; (2) the structure of the NRR (negative regulatory region), which is involved in sequestering the S2 cleavage site of Notch until interactions are formed with a DSL ligand; and (3) the structures of CSL, Notch, and Mastermind transcription complexes. In this section, we give a brief overview of the structures that have been determined and the functional information that was born out of these studies. For a more comprehensive review of this subject matter, please see Kovall and Blacklow (2010).
Both Notch and DSL are multidomain membrane proteins that contain a single transmembrane spanning region. The ectodomains of all Notch proteins contain multiple EGF repeats. LIN-12 has 13 repeats and GLP-1 has 10, in both cases fewer than found in the Drosophila (36) and mammalian Notch proteins (28-36). The EGF repeats of mammalian Notch proteins are heavily modified by O-linked glycans, and only a subset of these repeats is important for interacting with a DSL ligand. Similarly, the extracellular domains of DSL proteins also contain multiple EGF repeats, as well as the DSL domain that gives this family of ligands its name.
EGF repeats are small modular domains that typically contain three disulphide bonds and are primarily composed of β-strand, forming an extended rod-like structure. Some, but not all, EGF repeats bind calcium, which has been shown to be important for receptor-ligand complexation. Hanford and coworkers (Cordle et al., 2008) have determined several structures of EGF repeats using both NMR and crystallographic methods; these include (1) the EGF repeats 11-13 from human Notch1, which are necessary for interacting with ligand, and (2) the DSL domain followed by EGF repeats 1-3 from human Jagged1 (Figs. 3 and 4). While previous studies have shown that these regions of Notch1 and Jagged1 are important for receptor-ligand complexation and signaling in vivo, the isolated fragments interact only weakly in vitro (Cordle et al., 2008). Nonetheless, the EGF repeat structures revealed conserved interfaces on both Notch1 and Jagged1 that were shown to be important for both trans-signaling and cis-inhibition by in vivo assays in Drosophila (Cordle et al., 2008a; Cordle et al., 2008b). We note that the first two EGF-like motifs of Jagged1 correspond to the “DOS domain,” and that the structural information suggests that they are bona fide EGF repeats rather than a structurally distinctive domain.
Figure 3. EGF repeats structure from human Notch1. Ribbon diagram of EGF repeats 11-13 from human Notch1 (PDB ID: 2VJ3). Repeats 11, 12, and 13 are colored green, yellow, and magenta, respectively. Disulfide bonds are represented as grey sticks and bound calcium ions are cyan spheres.
Figure 4. Structure of the DSL domain and EGF repeats from the human ligand Jagged1. Ribbon diagram of DSL domain and EGF repeats 1-3 from human Jagged1 (PDB ID: 2VJ2). The DSL domain is colored yellow, and EGF repeats one, two, and three are colored cyan, green, and magenta, respectively. Disulfide bonds are shown as grey sticks.
The EGF repeats of Notch are followed by the NRR (negative regulatory region). The NRR functions to keep Notch in an off state until it interacts with a DSL ligand. Interestingly, constitutively activating mutations in the NRR were first identified in lin-12 and glp-1, and then later leukemia-causing mutations were found to cluster at the NRR of human Notch1; these mutations destabilize the structure of the NRR, causing the Notch receptor to be constitutively active (Greenwald and Seydoux, 1990; Berry et al., 1997; Pepper et al., 2003; Weng et al., 2004). Blacklow and coworkers determined the structure of the NRR in its auto-inhibited form (Figure 5) (Gordon et al., 2007; Gordon et al., 2009). It is composed of three LIN-12/Notch Repeat (LNR) domains and a heterodimerization domain (HD). Each LNR domain is disulphide bonded and binds calcium. Collectively, the tertiary structure is such that the three LNR domains protect the S2 site contained within the HD from cleavage. The structure of the NRR suggests that some sort of conformational change or unfolding event must occur to expose the S2 site for cleavage by ADAM; however, the molecular details of this process are unclear. One current model in the field is that endocytosis of the ligand-receptor complex in the signaling cell creates a mechanical force that exposes the NRR, or that an allosteric change occurs in the NRR upon formation of the DSL-Notch complex (reviewed in Kopan and Ilagan, 2009).
Figure 5. NRR structure from human Notch2. Ribbon diagram of the NRR from human Notch2 (PDB ID: 2OO4). LNR-A is colored green, LNR-B is cyan, and LNR-C is orange. Calcium and Zinc ions are represented as grey and blue spheres, respectively. The HD domain is colored magenta for the region N-terminal to the S1 cleavage site and yellow for the region C-terminal to the S1 cleavage site. The S2 cleavage site is colored red.
Perhaps the most extensive progress to date has been made in solving the structures of the proteins that are involved in regulating transcription from Notch target genes. These include structures of the DNA binding protein CSL, the RAM and ankyrin domains (ANK) of the receptor Notch, and the transcriptional coactivator Mastermind (MAM) (Kovall and Blacklow, 2010). Furthermore, several of these structures have been determined for components from C. elegans, Drosophila, and mammals, which illustrate the high degree of structural conservation between organisms. The first structure of CSL bound to a cognate DNA was determined using the C. elegans ortholog LAG-1 (Figure 6) (Kovall and Hendrickson, 2004). The structure revealed that all CSL proteins have a highly conserved core that is composed of three domains: an N-terminal domain (NTD), a β-trefoil domain (BTD), and a C-terminal domain (CTD). The NTD and CTD are Rel homology domains that are structurally similar to other DNA binding transcription factors, such as NF-kappaB and NFAT. The BTD derives its name from the pseudo three-fold arrangement of β-strands that compose this domain. The NTD and BTD of CSL form a continuous electropositive surface that makes specific contacts in the major and minor grooves, respectively, to recognize DNA (Kovall and Hendrickson, 2004). As discussed below in the context of CSL-NICD-MAM ternary complex structures, the BTD and CTD, as well as the NTD, make protein-protein interactions with transcriptional coregulators.
Figure 6. Structure of LAG-1 (CSL) bound to DNA. Ribbon diagram of CSL from C. elegans (PDB ID: 1TTU). The NTD, BTD, and CTD are colored cyan, green, and orange, respectively. A β-strand that bridges interactions with all three domains is colored magenta. The DNA is shown in a stick representation with carbon, oxygen, nitrogen, and phosphorous atoms colored yellow, red, blue, and orange, respectively.
Following the structure of LAG-1 bound to DNA, the structure of the transcriptionally active CSL-NICD-MAM ternary complex bound to DNA was determined for Notch components from both C. elegans (LAG-1/LIN-12/SEL-8) and humans (RBP-J/NICD1/MAML1) (Nam et al., 2006; Wilson and Kovall, 2006) (Figures 7, 8). Remarkably, despite the evolutionary sequence divergence between nematodes and mammals, the overall architecture of these two complexes is well conserved. The structures show that the RAM domain of NICD, which is intrinsically disordered prior to complexation (Nam et al., 2003; Bertagna et al., 2008), binds the BTD of CSL. The ANK domain of NICD, which primarily binds the CTD of CSL, consists of seven iterative ankyrin repeats, as well as a poorly conserved N-terminal capping repeat that was observed in the C. elegans complex structure. MAM adopts a strikingly bent helical conformation, which binds to a continuous groove formed by the CTD-ANK interface and the NTD of CSL. It should also be mentioned that there were several structural differences observed between the C. elegans and human CSL-NICD-MAM ternary complexes (Kovall, 2007). Most notable was the compact arrangement of domains observed in CSL for the C. elegans complex structure, but not the human structure. The functional significance of this observation has yet to be determined.
Figure 7. Structure of the C. elegans LAG-1/LIN-12/SEL-8 ternary complex structure bound to DNA. Ribbon diagram of the CSL-NICD-MAM ternary complex from C. elegans (PDB ID: 2FO1). CSL (LAG-1) and DNA are colored as in Figure 6. The RAM and ANK domains of NICD (LIN-12) are colored red and blue, respectively. MAM (SEL-8) is colored grey.
Figure 8. Structure of dimeric human CSL-NICD-MAM ternary complexes bound to HES-1 DNA. Figure shows two CSL-NICD-MAM ternary complexes bound to the HES-1 SPS for Notch components from humans. CSL, NICD, and MAM are colored green, blue, and red, respectively. The DNA is colored grey. A magenta asterisk denotes the site of cooperative interactions between the ANK domains of NICD.
A number of additional structures have been determined for the individual components, as well as complexes that represent assembly intermediates. These include Notch ANK structures from Drosophila and mammals (Zweifel et al., 2003; Nam et al., 2006), CSL-RAM-DNA complex structures from C. elegans (Friedmann et al., 2008), and a structure of murine CSL bound to DNA (Friedmann et al., 2008). In parallel, several biochemical and biophysical studies were published that provided a detailed mechanism for how the transcriptionally active CSL-NICD-MAM ternary complex assembles. The RAM domain of NICD forms a high affinity interaction with the BTD of CSL. This targets NICD to CSL and has three important functional ramifications: (1) RAM has been shown to be important for displacing transcriptional corepressors from CSL; (2) the tethering of NICD to CSL via RAM greatly increases the local concentration of ANK (Bertagna et al., 2008), which is necessary to facilitate the very weak CTD-ANK interaction (Del Bianco et al., 2008; Friedmann et al., 2008); and (3) it has been proposed that RAM induces a conformational change in CSL that facilitates assembly of the ternary complex. It should be mentioned, however, that the degree of allosteric coupling varied between Notch components from different organisms. Following the complexation of CSL-NICD, Mastermind binds the complex (Nam et al., 2003). Similar to the ANK domain of NICD, MAM also has very weak intrinsic affinity for either CSL or Notch, though when all three factors are present, a stable high affinity ternary complex is formed (Nam et al., 2003).
Another point worth noting is that originally it was thought that CSL had high affinity for DNA (~1nM Kd) and therefore was constitutively bound at target genes (Hamaguchi et al., 1989); thus, exchange of coregulators would take place on DNA-bound CSL. However, it was later determined that CSL only has moderate affinity for DNA (~100nM Kd), which is at least 10-fold weaker than the affinity of CSL for the RAM domain of NICD (Friedmann and Kovall, 2010). Interestingly, for both C. elegans and mammalian components, the ternary complex of CSL with NICD and MAM neither changes the specificity nor the affinity of CSL for DNA (Del Bianco et al., 2008; Friedmann et al., 2008). Furthermore, to date, none of the various corepressors that interact with CSL affect its affinity for DNA (Maier et al., 2011; VanderWielen et al., 2011). (Note: DIN-1 is the C. elegans ortholog of the mammalian corepressor MINT, but the Drosophila corepressor Hairless is not found in C. elegans). Taken together, these observations suggest that there may be some CSL-coregulator complexes in the nucleus that are bound to DNA and others that are free in the nucleoplasm. However, as all of these aforementioned binding studies were done with purified proteins produced in bacteria, the possibility that post-translational modifications affect the DNA binding properties of CSL cannot be excluded at this time.
LAG-1 appears to have the same binding site preference as its Drosophila and mammalian orthologs: LAG-1 protein binds to ATGGGAA in vitro, a sequence that is required in vivo for LIN-12 autoregulation (Wilkinson et al., 1994), and binding is specifically competed by the oligonucleotides containing the consensus RTGGGAA sequence (Christensen et al., 1996). Both YRTGRGAA and YGTGDGAA binding motifs have been identified in validated target genes in C. elegans.
In C. elegans, targets of the activation complex have been identified through forward computational screens based on the presence of binding site consensus sequences. Targets have also been recognized retrospectively by scanning genes with expression patterns or functional characteristics suggesting a role in Notch-mediated processes for the presence of potential binding sites. Showing that mutation of the potential binding sites abolishes expression can be taken as strong evidence that the gene is a direct target for the activation complex in vivo, as cell fate transformations in mutant backgrounds may confound the interpretation of a change in expression of a potential target. Table 2 summarizes target genes identified to date.
Table 2. Validated targets of LIN-12 or GLP-1
| Target gene | Context | Product | References |
|---|---|---|---|
| lin-12 | VU* | LIN-12/Notch | Wilkinson et al. 1994 |
| mir-61/250 | VU* | microRNA | Yoo and Greenwald, 2005 |
| lip-1 | VPC**, germline | ERK phosphatase | Berset et al., 2001; Lamont et al., 2004; Lee et al., 2006 |
| lst-1 | VPC**, germline | novel nematode-specific | Yoo et al., 2004; Singh et al., 2011 |
| lst-2 | VPC** | FYVE finger protein | Yoo et al., 2004 |
| lst-3 | VPC | SAP domain/DNA binding? | Yoo et al., 2004 |
| lst-4 | VPC | SNX9 sorting nexin | Yoo et al., 2004 |
| fbf-1 | germline | RNA binding protein | Lamont et al., 2004; Lee et al., 2006 |
| pkg-1 (egl-4) | neurons | cGMP-dependent protein kinase | Singh et al. 2004 |
|
*VU = Ventral Uterine precursor cell of the somatic gonad **VPC = Vulval Precursor Cell (specifically, P5.p and P7.p). A list of other potential targets, predicted computationally using the same crtieria that led to the validated targets lst-1-4, is given in the Supplemental Material of Yoo et al., 2004. |
|||
Interestingly, none of the identified C. elegans target genes contain paired CSL binding sites. Originally identified within the Enhancer of split locus in Drosophila and termed SPS (SuH paired site), an SPS consists of two CSL binding sites in a head-to-head arrangement separated by approximately 16 base pairs (Cave et al., 2005). In mammals, the well-established Notch target gene hes-1 contains a prototypical SPS (Jarriault et al., 1995). The SPS binds two CSL-NICD-MAM ternary complexes in a cooperative manner, which lowers the amount of NICD necessary to activate a target gene (Nam et al., 2007). Blacklow and colleagues determined the structure of dimeric human Notch transcription complexes bound at the hes-1 SPS promoter element (Arnett et al., 2010). The structure showed that the cooperativity was mediated by interactions between the ANK domains of NICD, which induced a modest bending in the DNA. The ANK residues required for cooperativity are not conserved in the C. elegans orthologs LIN-12 and GLP-1 (Nam et al., 2007), which raises the question as to whether the SPS motif is utilized in nematodes to regulate transcription from Notch target genes.
In C. elegans, genetic suppressor and enhancer screens have yielded candidate modulators as well as core components. Modulators are not essential for Notch signaling but can influence Notch activity. They may function generally or only in specific cellular contexts. They may act “directly”, through modifying or otherwise physically interacting with Notch, leading to altered Notch level, activity or stability. Or they may act “indirectly", by affecting cell physiology in ways that synergize with or antagonize Notch activity. The kind of modifiers identified seems heavily influence by the particular starting allele and the cellular context that form the basis of a given screen.
Here, we summarize some of the suppressor and enhancer screens that have been performed, starting with mutations that elevate or reduce Notch activity. Such screens have been done on large scales starting with specific alleles of the Notch genes lin-12 or glp-1, and with loss-of-function mutations in two components of γ-secretase, sel-12 and aph-1. The simple predictions for recovery of loss-of-function mutations in modulators in these starting conditions are given below, as well as a cross-reference to the section below in which such screens are detailed as in the table:
| starting condition | loss of a positive modulator | loss of a negative modulator |
|---|---|---|
| elevated Notch | suppress (section 6.1 below) | enhance (section 6.3) |
| reduced Notch | enhance (section 6.2) | suppress (section 6.4) |
Classifying a gene as a “modulator” rather than a core component is somewhat provisional. If a null allele of the gene results in a Notch phenotype, a good case can be made that it is a core component; however, maternal rescue (e.g., as for aph-1 and aph-2) or functional redundancy (e.g., as for sel-12 and hop-1) may obscure an essential role. Thus, a gene may shift from being classified as a “modulator” to a “core component” with further study. In addition, some modulators may be more general, i.e., function in multiple cellular contexts, whereas others may be more restricted. Some modulators may show allele-specificity within the same cellular context, which may be indicative of a role in an aspect of cell physiology or protein homeostasis rather than in Notch signaling per se. Thus, the category of “modulator” is quite heterogeneous and does not have distinct boundaries.
Genes provisionally classified as modulators through such genetic interactions are listed in conjunction with a brief description of the main screens used to identify them. Selected genes identified in candidate gene assays are also shown. Some modulators have been characterized in numerous assays; not all assays are shown for a given modulator, so for more details about individual genes please use the hyperlinks to Wormbase.
We note that we have not listed all of the genetic interactions associated with a given modulator. Furthermore, we note that even if a modulator is named after the screen in which it was identified, its function is not necessarily restricted to a particular context. For example, many genes described below are named ”sel” (suppressor/enhancer of lin-12), because their genetic behavior as a suppressor or enhancer depends on the nature of the lin-12 allele with which they are combined; furthermore, many sel genes also interact genetically with glp-1.
Suppressors of lin-12(d). The excess activity of lin-12(d) mutants prevents the specification of the anchor cell of the gonad, resulting in the absence of vulval induction and hence an egg-laying defect (see WormBook chapter Vulval development). Suppression of this defect has yielded null or hypomorphic mutations in many core components and positive modulators (Tax and Thomas, 1994; Katic et al., 2005) (Table 3) and neomorphic alleles of lag-2 (Tax and Thomas, 1994; Tax et al., 1997). In addition to lacking a functional vulva, “strong” lin-12(d) alleles also have a “Multivulva” phenotype because all six Vulval Precursor Cells (VPCs), instead of just two, adopt the particular fate resulting from LIN-12 activation (see Vulval development); suppression of this defect yielded sel-12 (Levitan and Greenwald, 1995).
Table 3. Suppressors of lin-12(d)
| Gene | Product | Drosophila | Mammals | Role/effect | Worm refs |
|---|---|---|---|---|---|
| lag-2 (sel-3)* | DSL protein | DSL | DSL | Ligand | (Tax and Thomas, 1994; Tax et al., 1997) |
| sel-5 | Kinase | NAK | AAK1 | modulate Notch endocytosis** | (Fares and Greenwald, 1999) |
| emb-4 (sel-6)** | nuclear ATPase | AAQ22465 | Aquarius | unknown | (Katic and Greenwald, 2006) |
| sel-7 | Novel, nuclear | n/a | n/a | unknown | (Chen et al., 2004) |
| sel-8 (lag-3)*** | Glutamine-rich | Mastermind | Mastermind | Nuclear complex | (Doyle et al., 2000) |
| sel-12 | Presenilin | Presenilin | Presenilin | g-secretase | (Levitan and Greenwald, 1995) |
| bre-5 | galactosyl-transferase | Brainiac | B3gnt4-001 | protein and lipid modification | (Katic et al., 2005) |
| lag-1*** | DNA binding protein | Su(H) | CBF1 | binding to target gene regulatory sequences | (Katic et al., 2005) |
|
Alternate, unofficial names are listed in parentheses. bold, core component *Suppressor alleles are neomorphic. **see (Gupta-Rossi et al., 2011) ***Suppressor alleles are partial loss-of-function. Unless otherwise designated, others are null alleles, although we note that sel-12 null mutants are viable because of functional redundancy with hop-1. |
|||||
Suppressors of glp-1 gain-of-function. glp-1(ar202) results in elevated glp-1 activity but remains significantly sensitive to ligand (Pepper et al., 2003). At 25oC, glp-1(ar202) mutants have ectopic proximal proliferation that results in sterility and a late-onset tumorous phenotype. This allele has both a complicated phenotype and complicated genetic behavior, which have consequences for the types of modifiers that interact with it, as discussed by Hubbard (2011).
Loss of either of two tetraspanin-encoding genes, tsp-12 and tsp-14, suppress glp-1(ar202) sterility; further characterization in C. elegans and human cell culture suggested that a subset of Tetraspanins modulate γ-secretase activity (Dunn et al., 2010). Null alleles of rsks-1, which encodes the sole worm homolog of mammalian p70S6K, also suppress glp-1(ar202) (Korta et al., 2012). Suppression of glp-1(ar202) has also been used to evaluate C. elegans orthologs of mammalian proteins found in physical association with γ-secretase; (M. Vargas and I. Greenwald, unpublished observations).
Enhancers of a glp-1 hypomorph. Five ego genes, for enhancers of glp-one, were identified by enhancing the partial germline proliferation defect of a weak glp-1 loss-of-function mutation (Qiao et al., 1995; Smardon et al., 2000). Three of these genes have been cloned, and additional candidate genes that display similar genetic interactions have also been identified (Table 4).
Table 4. Enhancers of a glp-1 hypomorph
| Gene | Product | Mammalian | Role/effect | Worm refs |
|---|---|---|---|---|
| ego-1 | RNA-directed RNA polymerase | n/a | many aspects of germline development | (Smardon et al., 2000) |
| ego-2 | Bro1-domain | protein trafficking | (Liu and Maine, 2007) | |
| atx-2 (ego-4) | Ataxin 2-like | Ataxin-2 | translational regulation | (Ciosk et al., 2004; Maine et al., 2004) |
| lag-1 | CSL | CBF1 | core component | (Qiao et al., 1995; Katic et al., 2005) |
| Candidate assay | ||||
| epn-1 | Epsin | Epsin | ligand endocytosis | (Tian et al., 2004) |
| csr-1 | nematode-specific Argonaut | n/a | many | (She et al., 2009) |
| ekl-1 | Tudor domain | n/a | many | (She et al., 2009) |
| drh-3 | DEAH/D box helicase | many | (She et al., 2009) | |
| cye-1 | Cyclin | IF1H1 | cell cycle regulation | (Fox et al., 2011) |
|
Alternate, unofficial names are listed in parentheses. bold, core component. Note that assigning epn-1 as a core component is provisional, as discussed in Section 3. |
||||
It is notable that EGO-2 is one of two Bro1-domain proteins in C. elegans; the other, ALX-1, was isolated in a yeast two-hybrid screen with the intracellular domain of LIN-12 and was found to be required for LIN-12 trafficking and degradation in the VPCs (Shaye and Greenwald, 2005) (see Section 6.3). However, ego-2 and alx-1 appear to have distinct roles: ego-2, like epn-1, appears to function mainly in the signaling cell (Tian et al., 2004; Liu and Maine, 2007), and ego-2 and alx-1 can either synergize or antagonize depending on context (Liu and Maine, 2007). The cellular focus of ego-2 and epn-1 and their interactions with glp-1 suggest that endocytosis of DSL proteins influences their activity in C. elegans, as in other organisms, despite unusual aspects of the structure.
Enhancers of reduced γ-secretase activity. Different synthetic phenotypes are obtained when null alleles of sel-12 and hop-1 are combined. The hop-1; sel-12 double mutants segregating from hop-1 heterozygous mothers display a Glp-1 sterile phenotype (Westlund et al., 1999). Screens for other genes that cause Glp-1 sterility in combination with sel-12 identified the novel gene pen-2 (“Presenilin ENhancer”) as well as the aph-1 and aph-2 genes that had been identified in a phenotype-based screen (Francis et al., 2002) (Table 1).
Enhancers of lin-12(d). sel-2 was found serendipitously in a screen for suppressors of the egg-laying defect of a weak lin-12(d) allele, but instead proved to be a strong enhancer in the VPCs: the activity of the weak allele is enhanced to result in the Multivulva phenotype associated with strong alleles (de Souza et al., 2007). SEL-2 is orthologous to mammalian Neurobeachin/LRBA. Loss of sel-2 activity affects the trafficking of LIN-12 in the VPCs and appears to affect trafficking in other polarized epithelial cells as well (de Souza et al., 2007).
Two other genes, alx-1 (see also Section 6.2) and wwp-1, which encodes the WWP-1/Su(dx)/Itch ubiquitin ligase, were found in a yeast two hybrid screen and found to facilitate LIN-12 trafficking and degradation in the VPCs (Shaye and Greenwald, 2005). Although these two genes were not assessed for their effects on lin-12(d) activity, aberrant trafficking of LIN-12 can lead to gain-of-function phenotypes (Shaye and Greenwald, 2005), a phenomenon also observed in Drosophila (e.g., Wilkin et al., 2004).
sel-10, identified as a suppressor of reduced lin-12 activity (see 6.4), also enhances weak lin-12(d) alleles to cause a Multivulva phenotype (Sundaram and Greenwald, 1993b; Hubbard et al., 1997). Ongoing screens have identified many additional genes that can enhance lin-12(d) activity (Y. Deng and I. Greenwald, unpublished observations).
Enhancers of glp-1 gain-of-function. Activation of glp-1 in the germ line promotes mitotic proliferation and a “tumorous” phenotype (Berry et al., 1997). A screen for enhancers of a weak tumorous phenotype caused by mild glp-1 gain-of-function identified four tumorous enhancers of glp-1 (teg genes), potential negative regulators of glp-1 activity in the germ line, three of which have been cloned (Table 5).
Table 5. Enhancers of glp-1 gain-of-function
| Gene | Product | Mammalian | Role/effect | Worm refs |
|---|---|---|---|---|
| teg-1 | splicing factor | CD2BP2 | pre-mRNA splicing | (Wang et al., 2012) |
| teg-4 | splicing factor | SAP130 | downstream of glp-1 | (Mantina et al., 2009) |
| prp-17* | splicing factor | PRP17 | downstream of glp-1 | (Kerins et al., 2010) |
| puf-8 (teg-2) | Pumilio-related | Pumilio homolog 2 | translational control | (Racher and Hansen, 2012) |
| pas-5 | proteasome subunit | PSMA5 | protein stability | (Macdonald et al., 2008) |
| mett-10** | methyltransferase | METTL16 | nuclear protein | (Dorsett et al., 2009) |
| him-17 | THAP domains | N/A | localizes to chromatin | Bessler et al., 2007 |
| Candidate assay | ||||
| dlc-1 | dynein light chain | DYNLL2 | dynein motor complex | (Dorsett and Schedl, 2009) |
| dhc-1 | dynein heavy chain | DNAH9 | dynein motor complex | (Dorsett and Schedl, 2009) |
|
Alternate, unofficial names are listed in parentheses. *see (Kerins et al., 2010) for a long list of many different splicing factor orthologs that show similar behavior. **mett-10 was originally identified by a rare Tumorous allele; loss of function mutations enhance a weak tumorous allele of glp-1. DLC-1 and DHC-2 interact with METT-10 in a yeast two-hybrid assay (discussed in Dorsett and Schedl, 2009). |
||||
Suppressors of mutations that reduce γ-secretase activity. Two different screens have been done in this category. One was based on the egg-laying defect of sel-12 null mutants, which cannot lay eggs because of a lin-12-mediated gonadal defect (Cinar et al., 2001), but are viable because they retain hop-1/Presenilin activity. Suppressors of the egg-laying defect of sel-12 mutants identified null or hypomorphic alleles of five spr genes (suppressors of presenilin) (Wen et al., 2000). One gene, spr-2, encodes the ortholog of the likely chromatin factor SET (Wen et al., 2000). Four genes appear to act together: spr-1/CoREST and spr-5/LSD1, members of the mammalian CoREST co-repressor complex, and spr-3 and spr-4, which encode two zinc-finger proteins that may function in a complex with CoREST (Eimer et al., 2002; Jarriault and Greenwald, 2002; Lakowski et al., 2003). Loss of CoREST activity has been proposed to result in increased expression of hop-1 (Eimer et al., 2002; Lakowski et al., 2003) or an altered balance of repression and activation of lin-12 target genes (Jarriault and Greenwald, 2002). Interestingly, in murine T-ALL cells, there may be some association between the binding sites for CSL and REST, a transcription factor that recruits CoREST to repress target genes (Wang et al., 2011).
Another screen in this category was based on suppression of the maternal effect lethal phenotype of a hypomorphic allele of the aph-1 component of γ-secretase (Hale et al., 2012). This screen identified sao-1, which physically interacts with SEL-10. SEL-10 is an E3 ubiquitin ligase that directly affects the stability of LIN-12 and GLP-1 (Hubbard et al., 1997; Welcker and Clurman, 2008) and has also been implicated in SEL-12/Presenilin stability (Wu et al., 1998). sao-1 displays numerous additional genetic interactions with other components of the Notch signaling system, all consistent with the proposal that SAO-1 may act with SEL-10 in downregulating one or more components that influence Notch activity (Hale et al., 2012).
Suppressors of lin-12 hypomorphic alleles. Partial loss-of-function (hypomorphic) mutants of lin-12 have a functional vulva but are egg-laying defective (Sundaram and Greenwald, 1993a). Suppression of the egg-laying defect has been used as the basis of a screen for suppressors (Sundaram and Greenwald, 1993b) and to characterize other interactors (Table 6).
Table 6. Suppressors of lin-12 hypomorphs
| Gene | Product | Mammalian | Role/effect | Worm refs |
|---|---|---|---|---|
| sel-1 | Extracellular, GPI-linked | HRD3, SEL1L | Unfolded Protein Response | (Grant and Greenwald, 1996, 1997) |
| sel-9 | p24 ER protein | p24 | ER quality control | (Wen and Greenwald, 1999) |
| sel-10 | E3 ubiquitin ligase | Fbw7 | Directs turnover of Notch(intra) | (Hubbard et al., 1997) |
| sel-11 (hrd-1) | E3 ubiquitin ligase | HRD1 | Unfolded Protein Response | (Choi et al., 2010) |
|
Alternate, unofficial names are listed in parentheses. *For null alleles of sel-10 to suppress the egg-laying defect of lin-12(n676n930), an as-yet-uncloned second mutation, arX, must be present (Sundaram and Greenwald, 1993b). |
||||
sel-1, sel-9, and sel-11 exert their effects through physiological processes. In contrast, SEL-10 directly binds to the LIN-12 intracellular domain through a “Cdc4 phosphodegron” sequence and regulates its stability (Hubbard et al., 1997; de la Cova and Greenwald, 2012). This role is conserved in the mammalian ortholog, Fbw7, which also acts as a tumor suppressor in many contexts (Welcker and Clurman, 2008). In particular, Fbw7 function as a tumor suppressor appears to be mediated through its effect on Notch in T-ALL (Thompson et al., 2007).
Suppressors of glp-1 hypomorphic alleles. Screens for suppressors of the germline defect caused by loss of zygotic glp-1 activity (Maine and Kimble, 1993) or of the embryonic lethality caused by loss of maternal glp-1 activity (A.M. Howell and J. Priess, personal communication) identified ten sog genes, for suppressor of glp-1. None of these have been molecularly characterized. Additional suppressors of glp-1sterility proved to be in genes required for body morphology, which may indicate that the extracellular matrix influences glp-1 activity in the germ line (Maine and Kimble, 1989).
Studies of lin-12 and glp-1 in C. elegans have continued to yield new insights into the Notch signaling system and its regulation. Between now and the next update, we expect to see further progress towards a structural understanding of Notch signaling; potentially, predictions from structural studies could be tested in vivo in C. elegans. We also expect new modulators to be defined through genetic screens, and that the human orthologs of some of these modulators will be connected to human disease processes.
We thank Caroline Goutte, Jane Hubbard, Eleanor Maine, and members of the Greenwald and Kovall laboratories for comments on the manuscript. Work in the Greenwald lab was supported by a grant from the NIH (CA095389) and by the Howard Hughes Medical Institute. Research in the Kovall lab was supported by grants from the NIH (CA120199), the Leukemia & Lymphoma Society, and the Muscular Dystrophy Association.
Arnett, K.L., Hass, M., McArthur, D.G., Ilagan, M.X., Aster, J.C., Kopan, R., and Blacklow, S.C. (2010). Structural and mechanistic insights into cooperative assembly of dimeric Notch transcription complexes. Nat. Struct. Mol. Biol. 17, 1312-1317. Abstract Article
Austin, J., and Kimble, J. (1987). glp-1 is required in the germ line for regulation of the decision between mitosis and meiosis in C. elegans. Cell 51, 589-599. Abstract Article
Berry, L.W., Westlund, B., and Schedl, T. (1997). Germ-line tumor formation caused by activation of glp-1, a Caenorhabditis elegans member of the Notch family of receptors. Development 124, 925-936. Abstract
Berset, T., Holer E.F., Battu, G., Canevascini, S., and Hajnal, A. (2001). Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 291, 1055-1058. Abstract Article
Bertagna, A., Toptygin, D., Brand, L., and Barrick, D. (2008). The effects of conformational heterogeneity on the binding of the Notch intracellular domain to effector proteins: a case of biologically tuned disorder. Biochem. Soc. Trans. 36, 157-166. Abstract Article
Bessler, J.B., Reddy, K.C., Hayashi, M., Hodgkin, J. and Villeneuve, A.M. (2007) A role for Caenorhabditis elegans chromatin-associated protein HIM-17 in the proliferation vs. meiotic entry decision. Genetics 175, 2029-2037. Abstract Article
Blaumueller, C.M., Qi, H., Zagouras, P., and Artavanis-Tsakonas, S. (1997). Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 90, 281-291. Abstract Article
Brou, C., Logeat, F., Gupta, N., Bessia, C., LeBail, O., Doedens, J.R., Cumano, A., Roux, P., Black, R.A., and Israel, A. (2000). A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol. Cell 5, 207-216. Abstract Article
Cave, J.W., Loh, F., Surpris, J.W., Xia, L., and Caudy, M.A. (2005). A DNA transcription code for cell-specific gene activation by notch signaling. Curr. Biol. 15, 94-104. Abstract Article
Chen, J., Li, X., and Greenwald, I. (2004). sel-7, a positive regulator of lin-12 activity, encodes a novel nuclear protein in Caenorhabditis elegans. Genetics 166, 151-160. Abstract Article
Chen, N., and Greenwald, I. (2004). The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev. Cell 6, 183-192. Abstract Article
Choi, M.S., Yoo, A.S., and Greenwald, I. (2010). sel-11 and cdc-42, two negative modulators of LIN-12/Notch activity in C. elegans. PLoS One 5, e11885. Abstract Article
Christensen, S., Kodoyianni, V., Bosenberg, M., Friedman, L., and Kimble, J. (1996). lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development 122, 1373-1383. Abstract
Cinar, H.N., Sweet, K.L., Hosemann, K.E., Earley, K., and Newman, A.P. (2001). The SEL-12 presenilin mediates induction of the Caenorhabditis elegans uterine pi cell fate. Dev. Biol. 237, 173-182. Abstract Article
Ciosk, R., DePalma, M., and Priess, J.R. (2004). ATX-2, the C. elegans ortholog of ataxin 2, functions in translational regulation in the germline. Development 131, 4831-4841. Abstract Article
Cordle, J., Johnson, S., Tay, J.Z., Roversi, P., Wilkin, M.B., de Madrid, B.H., Shimizu, H., Jensen, S., Whiteman, P., Jin, B., et al. (2008a). A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat. Struct. Mol. Biol. 15, 849-857. Abstract Article
Cordle, J., Redfieldz, C., Stacey, M., van der Merwe, P.A., Willis, A.C., Champion, B.R., Hambleton, S., and Handford, P.A. (2008b). Localization of the Delta-like-1-binding site in human Notch-1 and its modulation by calcium affinity. J. Biol. Chem. 283, 11785-11793. Abstract Article
Crittenden, S.L., Troemel, E.R., Evans, T.C., and Kimble, J. (1994). GLP-1 is localized to the mitotic region of the C. elegans germ line. Development 120, 2901-2911. Abstract
de la Cova, C., and Greenwald, I. (2012). SEL-10/Fbw7-dependent negative feedback regulation of LIN-45/Braf signaling in C. elegans via a conserved phosphodegron. Genes Dev. 26, 2524-2535. Abstract
de Souza, N., Vallier, L.G., Fares, H., and Greenwald, I. (2007). SEL-2, the C. elegans neurobeachin/LRBA homolog, is a negative regulator of lin-12/Notch activity and affects endosomal traffic in polarized epithelial cells. Development 134, 691-702. Abstract Article
Del Bianco, C., Aster, J.C., and Blacklow, S.C. (2008). Mutational and energetic studies of Notch 1 transcription complexes. J. Mol. Biol. 376, 131-140. Abstract Article
Dorsett, M., and Schedl, T. (2009). A role for dynein in the inhibition of germ cell proliferative fate. Mol. Cell. Biol. 29, 6128-6139. Abstract Article
Dorsett, M., Westlund, B., and Schedl, T. (2009). METT-10, a putative methyltransferase, inhibits germ cell proliferative fate in Caenorhabditis elegans. Genetics 183, 233-247. Abstract Article
Doyle, T.G., Wen, C., and Greenwald, I. (2000). SEL-8, a nuclear protein required for LIN-12 and GLP-1 signaling in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 97, 7877-7881. Abstract Article
Dunn, C.D., Sulis, M.L., Ferrando, A.A., and Greenwald, I. (2010). A conserved tetraspanin subfamily promotes Notch signaling in Caenorhabditis elegans and in human cells. Proc. Natl. Acad. Sci. U. S. A. 107, 5907-5912. Abstract Article
Edbauer, D., Winkler, E., Regula, J.T., Pesold, B., Steiner, H., and Haass, C. (2003). Reconstitution of γ-secretase activity. Nat. Cell Biol. 5, 486-488. Abstract Article
Eimer, S., Lakowski, B., Donhauser, R., and Baumeister, R. (2002). Loss of spr-5 bypasses the requirement for the C.elegans presenilin sel-12 by derepressing hop-1. EMBO J. 21, 5787-5796. Abstract Article
Fares, H., and Greenwald, I. (1999). SEL-5, a serine/threonine kinase that facilitates lin-12 activity in Caenorhabditis elegans. Genetics 153, 1641-1654. Abstract
Ferguson, E.L., and Horvitz, H.R. (1985). Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics 110, 17-72. Abstract
Fitzgerald, K., and Greenwald, I. (1995). Interchangeability of Caenorhabditis elegans DSL proteins and intrinsic signalling activity of their extracellular domains in vivo. Development 121, 4275-4282. Abstract
Fitzgerald, K., Wilkinson, H.A., and Greenwald, I. (1993). glp-1 can substitute for lin-12 in specifying cell fate decisions in Caenorhabditis elegans. Development 119, 1019-1027. Abstract
Fox, P.M., Vought, V.E., Hanazawa, M., Lee, M.H., Maine, E.M., and Schedl, T. (2011). Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development 138, 2223-2234. Abstract Article
Francis, R., McGrath, G., Zhang, J., Ruddy, D.A., Sym, M., Apfeld, J., Nicoll, M., Maxwell, M., Hai, B., Ellis, M.C., et al. (2002). aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev. Cell 3, 85-97. Abstract Article
Friedmann, D.R., and Kovall, R.A. (2010). Thermodynamic and structural insights into CSL-DNA complexes. Protein Sci. 19, 34-46. Abstract Article
Friedmann, D.R., Wilson, J.J., and Kovall, R.A. (2008). RAM-induced allostery facilitates assembly of a notch pathway active transcription complex. J. Biol. Chem. 283, 14781-14791. Abstract Article
Fryer, C.J., White, J.B., and Jones, K.A. (2004). Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16, 509-520. Abstract Article
Gordon, W.R., Roy, M., Vardar-Ulu, D., Garfinkel, M., Mansour, M.R., Aster, J.C., and Blacklow, S.C. (2009). Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood 113, 4381-4390. Abstract Article
Gordon, W.R., Vardar-Ulu, D., Histen, G., Sanchez-Irizarry, C., Aster, J.C., and Blacklow, S.C. (2007). Structural basis for autoinhibition of Notch. Nat. Struct. Mol. Biol. 14, 295-300. Abstract Article
Goutte, C., Hepler, W., Mickey, K.M., and Priess, J.R. (2000). aph-2 encodes a novel extracellular protein required for GLP-1-mediated signaling. Development 127, 2481-2492. Abstract
Goutte, C., Tsunozaki, M., Hale, V.A., and Priess, J.R. (2002). APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. U. S. A. 99, 775-779. Abstract Article
Grant, B., and Greenwald, I. (1996). The Caenorhabditis elegans sel-1 gene, a negative regulator of lin-12 and glp-1, encodes a predicted extracellular protein. Genetics 143, 237-247. Abstract
Grant, B., and Greenwald, I. (1997). Structure, function, and expression of SEL-1, a negative regulator of LIN-12 and GLP-1 in C. elegans. Development 124, 637-644. Abstract
Greenwald, I. (2005). LIN-12/Notch signaling in C. elegans. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.10.1, http://www.wormbook.org. Article
Greenwald, I. (2012). Notch and the awesome power of genetics. Genetics 191, 655-669. Abstract Article
Greenwald, I., and Seydoux, G. (1990). Analysis of gain-of-function mutations of the lin-12 gene of Caenorhabditis elegans. Nature 346, 197-199. Abstract Article
Greenwald, I.S., Sternberg, P.W., and Horvitz, H.R. (1983). The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell 34, 435-444. Abstract Article
Gupta-Rossi, N., Ortica, S., Meas-Yedid, V., Heuss, S., Moretti, J., Olivo-Marin, J.C., and Israel, A. (2011). The adaptor-associated kinase 1, AAK1, is a positive regulator of the Notch pathway. J. Biol. Chem. 286, 18720-18730. Abstract Article
Hale, V.A., Guiney, E.L., Goldberg, L.Y., Haduong, J.H., Kwartler, C.S., Scangos, K.W., and Goutte, C. (2012). Notch signaling is antagonized by SAO-1, a novel GYF-domain protein that interacts with the E3 ubiquitin ligase SEL-10 in Caenorhabditis elegans. Genetics 190, 1043-1057. Abstract Article
Hamaguchi Y., Matsunami N., Yamamoto Y., and Honjo T. (1989). Purification and characterization of a protein that binds to the recombination signal sequence of the immunoglobulin J kappa segment. Nucleic Acids Res. 17, 9015-9026. Abstract Article
Hobert O. (2010). Neurogenesis in the nematode Caenorhabditis elegans. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.12.2, http://www.wormbook.org. Article
Hoyos, E., Kim, K., Milloz, J., Barkoulas, M., Penigault, J.B., Munro, E., and Felix, M.A. (2011). Quantitative variation in autocrine signaling and pathway crosstalk in the Caenorhabditis vulval network. Curr. Biol. 21, 527-538. Abstract Article
Hubbard, E.J., Wu, G., Kitajewski, J., and Greenwald, I. (1997). sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes Dev. 11, 3182-3193. Abstract Article
Hubbard, E.J. (2011) Insulin and germline proliferation in Caenorhabditis elegans. Vitam. Horm. 87, 61-77. Abstract Article
Iwatsubo, T. (2004). The γ-secretase complex: machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 14, 379-383. Abstract Article
Jarriault, S., Brou, C., Logeat, F., Schroeter, E.H., Kopan, R., and Israel, A. (1995). Signalling downstream of activated mammalian Notch. Nature 377, 355-358. Abstract Article
Jarriault, S., and Greenwald, I. (2002). Suppressors of the egg-laying defective phenotype of sel-12 presenilin mutants implicate the CoREST corepressor complex in LIN-12/Notch signaling in C. elegans. Genes Dev. 16, 2713-2728. Abstract Article
Jarriault, S., and Greenwald, I. (2005). Evidence for functional redundancy between C. elegans ADAM proteins SUP-17/Kuzbanian and ADM-4/TACE. Dev. Biol. 287, 1-10. Abstract Article
Jarriault, S., Schwab, Y., and Greenwald, I. (2008). A Caenorhabditis elegans model for epithelial-neuronal transdifferentiation. Proc. Natl. Acad. Sci. U. S. A. 105, 3790-3795. Abstract Article
Katic, I., and Greenwald, I. (2006). EMB-4: a predicted ATPase that facilitates lin-12 activity in Caenorhabditis elegans. Genetics 174, 1907-1915. Abstract Article
Katic, I., Vallier, L.G., and Greenwald, I. (2005). New positive regulators of lin-12 activity in Caenorhabditis elegans include the BRE-5/Brainiac glycosphingolipid biosynthesis enzyme. Genetics 171, 1605-1615. Abstract Article
Kerins, J.A., Hanazawa, M., Dorsett, M., and Schedl, T. (2010). PRP-17 and the pre-mRNA splicing pathway are preferentially required for the proliferation versus meiotic development decision and germline sex determination in Caenorhabditis elegans. Dev. Dyn. 239, 1555-1572. Abstract Article
Kidd, S., and Lieber, T. (2002). Furin cleavage is not a requirement for Drosophila Notch function. Mech. Dev. 115, 41-51. Abstract Article
Kimble, J., and Crittenden, S.L. (2005). Germline proliferation and its control. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.13.1, http://www.wormbook.org. Article
Kitagawa, M., Oyama, T., Kawashima, T., Yedvobnick, B., Kumar, A., Matsuno, K., and Harigaya, K. (2001). A human protein with sequence similarity to Drosophila mastermind coordinates the nuclear form of notch and a CSL protein to build a transcriptional activator complex on target promoters. Mol. Cell. Biol. 21, 4337-4346. Abstract Article
Komatsu, H., Chao, M.Y., Larkins-Ford, J., Corkins, M.E., Somers, G.A., Tucey, T., Dionne, H.M., White, J.Q., Wani, K., Boxem, M., et al. (2008). OSM-11 facilitates LIN-12 Notch signaling during Caenorhabditis elegans vulval development. PLoS Biol 6, e196. Abstract Article
Kopan, R., and Ilagan, M.X. (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216-233. Abstract Article
Korta, D. Z., Tuck, S. and Hubbard, E.J. (2012) S6K links cell fate, cell cycle and nutrient response in C. elegans germline stem/progenitor cells. Development 139, 859-870. Abstract Article
Kostas, S.A., and Fire, A. (2002). The T-box factor MLS-1 acts as a molecular switch during specification of nonstriated muscle in C. elegans. Genes Dev. 16, 257-269. Abstract Article
Kovall, R.A. (2007). Structures of CSL, Notch and Mastermind proteins: piecing together an active transcription complex. Curr. Opin. Struc. Biol. 17, 117-127. Abstract Article
Kovall, R.A., and Blacklow, S.C. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr. Top. Dev. Biol. 92, 31-71. Article
Kovall, R.A., and Blacklow, S.C. (2010). Mechanistic insights into notch receptor signaling from structural and biochemical studies. Curr. Top. Dev. Biol. 92, 31-71. Abstract Article
Kovall, R.A., and Hendrickson, W.A. (2004). Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. EMBO J. 23, 3441-3451. Abstract Article
Lakowski, B., Eimer, S., Gobel, C., Bottcher, A., Wagler, B., and Baumeister, R. (2003). Two suppressors of sel-12 encode C2H2 zinc-finger proteins that regulate presenilin transcription in Caenorhabditis elegans. Development 130, 2117-2128. Abstract Article
Lambie, E.J., and Kimble, J. (1991). Two homologous regulatory genes, lin-12 and glp-1, have overlapping functions. Development 112, 231-240. Abstract
Lamont, L.B., Crittenden, S.L., Bernstein, D., Wickens, M., and Kimble, J. (2004). FBF-1 and FBF-2 regulate the size of the mitotic region in the C. elegans germline. Dev. Cell 7, 697-707. Abstract Article
LaRue, B.L., and Padilla, P.A. (2011). Environmental and genetic preconditioning for long-term anoxia responses requires AMPK in Caenorhabditis elegans. PLoS One 6, e16790. Abstract Article
Lee, M.-H., and Schedl, T. (2006). RNA-binding proteins. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.79.1, http://www.wormbook.org. Abstract Article
Levitan, D., Doyle, T.G., Brousseau, D., Lee, M.K., Thinakaran, G., Slunt, H.H., Sisodia, S.S., and Greenwald, I. (1996). Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 93, 14940-14944. Abstract Article
Levitan, D., and Greenwald, I. (1995). Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer's disease gene. Nature 377, 351-354. Abstract Article
Levitan, D., Yu, G., St. George Hyslop, P., and Goutte, C. (2001). APH-2/nicastrin functions in LIN-12/Notch signaling in the Caenorhabditis elegans somatic gonad. Dev. Biol. 240, 654-661. Abstract Article
Li, X., and Greenwald, I. (1996). Membrane topology of the C. elegans SEL-12 presenilin. Neuron 17, 1015-1021. Abstract Article
Li, X., and Greenwald, I. (1997). HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc. Natl. Acad. Sci. U. S. A. 94, 12204-12209. Abstract Article
Li, X., and Greenwald, I. (1998). Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc. Natl. Acad. Sci. U. S. A. 95, 7109-7114. Abstract Article
Liu, Y., and Maine, E.M. (2007). The Bro1-domain protein, EGO-2, promotes Notch signaling in Caenorhabditis elegans. Genetics 176, 2265-2277. Abstract Article
Logeat, F., Bessia, C., Brou, C., LeBail, O., Jarriault, S., Seidah, N.G., and Israel, A. (1998). The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. U. S. A. 95, 8108-8112. Abstract Article
Macdonald, L.D., Knox, A., and Hansen, D. (2008). Proteasomal regulation of the proliferation vs. meiotic entry decision in the Caenorhabditis elegans germ line. Genetics 180, 905-920. Abstract Article
Maier, D., Kurth, P., Schulz, A., Russell, A., Yuan, Z., Gruber, K., Kovall, R.A., and Preiss, A. (2011). Structural and functional analysis of the repressor complex in the Notch signaling pathway of Drosophila melanogaster. Mol. Biol. Cell 22, 3242-3252. Abstract Article
Maine, E.M., Hansen, D., Springer, D., and Vought, V.E. (2004). Caenorhabditis elegans atx-2 promotes germline proliferation and the oocyte fate. Genetics 168, 817-830. Abstract Article
Maine, E.M., and Kimble, J. (1989). Identification of genes that interact with glp-1, a gene required for inductive cell interactions in Caenorhabditis elegans. Development 106, 133-143. Abstract
Maine, E.M., and Kimble, J. (1993). Suppressors of glp-1, a gene required for cell communication during development in Caenorhabditis elegans, define a set of interacting genes. Genetics 135, 1011-1022. Abstract
Mango, S.E., Maine, E.M., and Kimble, J. (1991). Carboxy-terminal truncation activates glp-1 protein to specify vulval fates in Caenorhabditis elegans. Nature 352, 811-815. Abstract Article
Mango, S.E., Thorpe, C.J., Martin, P.R., Chamberlain, S.H., and Bowerman, B. (1994). Two maternal genes, apx-1 and pie-1, are required to distinguish the fates of equivalent blastomeres in the early Caenorhabditis elegans embryo. Development 120, 2305-2315. Abstract
Mantina, P., MacDonald, L., Kulaga, A., Zhao, L., and Hansen, D. (2009). A mutation in teg-4, which encodes a protein homologous to the SAP130 pre-mRNA splicing factor, disrupts the balance between proliferation and differentiation in the C. elegans germ line. Mech. Dev. 126, 417-429. Abstract Article
Mello, C.C., Draper, B.W., and Priess, J.R. (1994). The maternal genes apx-1 and glp-1 and establishment of dorsal-ventral polarity in the early C. elegans embryo. Cell 77, 95-106. Abstract Article
Mickey, K.M., Mello, C.C., Montgomery, M.K., Fire, A., and Priess, J.R. (1996). An inductive interaction in 4-cell stage C. elegans embryos involves APX-1 expression in the signalling cell. Development 122, 1791-1798. Abstract
Nadarajan, S., Govindan, J.A., McGovern, M., Hubbard, E.J., and Greenstein, D. (2009). MSP and GLP-1/Notch signaling coordinately regulate actomyosin-dependent cytoplasmic streaming and oocyte growth in C. elegans. Development 136, 2223-2234. Abstract Article
Nam, Y., Sliz, P., Pear, W.S., Aster, J.C., and Blacklow, S.C. (2007). Cooperative assembly of higher-order Notch complexes functions as a switch to induce transcription. Proc. Natl. Acad. Sci. U. S. A. 104, 2103-2108. Abstract Article
Nam, Y., Sliz, P., Song, L., Aster, J.C., and Blacklow, S.C. (2006). Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes. Cell 124, 973-983. Abstract Article
Nam, Y., Weng, A.P., Aster, J.C., and Blacklow, S.C. (2003). Structural requirements for assembly of the CSL·intracellular Notch1·Mastermind-like 1 transcriptional activation complex. J. Biol. Chem. 278, 21232-21239. Abstract Article
Ouellet, J., Li, S., and Roy, R. (2008). Notch signalling is required for both dauer maintenance and recovery in C. elegans. Development 135, 2583-2592. Abstract Article
Pan, D., and Rubin, G.M. (1997). Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell 90, 271-280. Abstract Article
Pepper, A.S., Killian, D.J., and Hubbard, E.J. (2003). Genetic analysis of Caenorhabditis elegans glp-1 mutants suggests receptor interaction or competition. Genetics 163, 115-132. Abstract
Petcherski, A.G., and Kimble, J. (2000a). LAG-3 is a putative transcriptional activator in the C. elegans Notch pathway. Nature 405, 364-368. Abstract
Petcherski, A.G., and Kimble, J. (2000b). Mastermind is a putative activator for Notch. Curr. Biol. 10, R471-473. Abstract Article
Priess, J.R. (2005). Notch signaling in the C. elegans embryo. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.4.1, http://www.wormbook.org. Abstract Article
Priess, J.R., Schnabel, H., and Schnabel, R. (1987). The glp-1 locus and cellular interactions in early C. elegans embryos. Cell 51, 601-611. Abstract Article
Qiao, L., Lissemore, J.L., Shu, P., Smardon, A., Gelber, M.B., and Maine, E.M. (1995). Enhancers of glp-1, a gene required for cell-signaling in Caenorhabditis elegans, define a set of genes required for germline development. Genetics 141, 551-569. Abstract
Racher, H., and Hansen, D. (2012). PUF-8, a Pumilio homolog, inhibits the proliferative fate in the Caenorhabditis elegans germline. G3 2, 1197-1205. Abstract Article
Rasmussen, J.P., English, K., Tenlen, J.R., and Priess, J.R. (2008). Notch signaling and morphogenesis of single-cell tubes in the C. elegans digestive tract. Dev. Cell 14, 559-569. Abstract Article
Rooke, J., Pan, D., Xu, T., and Rubin, G.M. (1996). KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science 273, 1227-1231. Abstract Article
Shaye, D.D., and Greenwald, I. (2005). LIN-12/Notch trafficking and regulation of DSL ligand activity during vulval induction in Caenorhabditis elegans. Development 132, 5081-5092. Abstract Article
She, X., Xu, X., Fedotov, A., Kelly, W.G., and Maine, E.M. (2009). Regulation of heterochromatin assembly on unpaired chromosomes during Caenorhabditis elegans meiosis by components of a small RNA-mediated pathway. PLoS Genet 5, e1000624. Abstract Article
Singh, K., Chao, M.Y., Somers, G.A., Komatsu, H., Corkins, M.E., Larkins-Ford, J., Tucey, T., Dionne, H.M., Walsh, M.B., Beaumont, E.K., et al. (2011). C. elegans Notch signaling regulates adult chemosensory response and larval molting quiescence. Curr. Biol. 21, 825-834. Abstract Article
Smardon, A., Spoerke, J.M., Stacey, S.C., Klein, M.E., Mackin, N., and Maine, E.M. (2000). EGO-1 is related to RNA-directed RNA polymerase and functions in germ-line development and RNA interference in C. elegans. Curr. Biol. 10, 169-178. Abstract Article
Sternberg, P.W. (2005). Vulval development. WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.6.1, http://www.wormbook.org. Article
Struhl, G., and Adachi, A. (2000). Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 6, 625-636. Abstract Article
Struhl, G., Fitzgerald, K., and Greenwald, I. (1993). Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell 74, 331-345. Abstract Article
Sundaram, M., and Greenwald, I. (1993a). Genetic and phenotypic studies of hypomorphic lin-12 mutants in Caenorhabditis elegans. Genetics 135, 755-763. Abstract
Sundaram, M., and Greenwald, I. (1993b). Suppressors of a lin-12 hypomorph define genes that interact with both lin-12 and glp-1 in Caenorhabditis elegans. Genetics 135, 765-783. Abstract
Tax, F.E., and Thomas, J.H. (1994). Cell-cell interactions. Receiving signals in the nematode embryo. Curr. Biol. 4, 914-916. Abstract Article
Tax, F.E., Thomas, J.H., Ferguson, E.L., and Horvitz, H.R. (1997). Identification and characterization of genes that interact with lin-12 in Caenorhabditis elegans. Genetics 147, 1675-1695. Abstract
Thinakaran, G., Borchelt, D.R., Lee, M.K., Slunt, H.H., Spitzer, L., Kim, G., Ratovitsky, T., Davenport, F., Nordstedt, C., Seeger, M., et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181-190. Abstract Article
Thompson, B.J., Buonamici, S., Sulis, M.L., Palomero, T., Vilimas, T., Basso, G., Ferrando, A., and Aifantis, I. (2007). The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 204, 1825-1835. Abstract Article
Tian, X., Hansen, D., Schedl, T., and Skeath, J.B. (2004). Epsin potentiates Notch pathway activity in Drosophila and C. elegans. Development 131, 5807-5815. Abstract Article
VanderWielen, B.D., Yuan, Z., Friedmann, D.R., and Kovall, R.A. (2011). Transcriptional repression in the Notch pathway: thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J. Biol. Chem. 286, 14892-14902. Abstract Article
Wang, C., Wilson-Berry, L., Schedl, T., and Hansen, D. (2012). TEG-1 CD2BP2 regulates stem cell proliferation and sex determination in the C. elegans germ line and physically interacts with the UAF-1 U2AF65 splicing factor. Dev. Dyn. 241, 505-521. Abstract Article
Wang, H., Zou, J., Zhao, B., Johannsen, E., Ashworth, T., Wong, H., Pear, W.S., Schug, J., Blacklow, S.C., Arnett, K.L., et al. (2011). Genome-wide analysis reveals conserved and divergent features of Notch1/RBPJ binding in human and murine T-lymphoblastic leukemia cells. Proc. Natl. Acad. Sci. U. S. A. 108, 14908-14913. Abstract Article
Wang, W., and Struhl, G. (2004). Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development 131, 5367-5380. Abstract Article
Welcker, M. and Clurman, B.E. (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 93-93. Abstract Article
Wen, C., and Greenwald, I. (1999). p24 proteins and quality control of LIN-12 and GLP-1 trafficking in Caenorhabditis elegans. J. Cell Biol. 145, 1165-1175. Abstract Article
Wen, C., Levitan, D., Li, X., and Greenwald, I. (2000). spr-2, a suppressor of the egg-laying defect caused by loss of sel-12 presenilin in Caenorhabditis elegans, is a member of the SET protein subfamily. Proc. Natl. Acad. Sci. U. S. A. 97, 14524-14529. Abstract
Wen, C., Metzstein, M.M., and Greenwald, I. (1997). SUP-17, a Caenorhabditis elegans ADAM protein related to Drosophila KUZBANIAN, and its role in LIN-12/NOTCH signalling. Development 124, 4759-4767. Abstract
Weng, A.P., Ferrando, A.A., Lee, W., Morris, J.P. 4th, Silverman, L.B., Sanchez-Irizarry, C., Blacklow, S.C., Look, A.T., and Aster, J.C. (2004). Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269-271. Abstract Article
Westlund, B., Parry, D., Clover, R., Basson, M., and Johnson, C.D. (1999). Reverse genetic analysis of Caenorhabditis elegans presenilins reveals redundant but unequal roles for sel-12 and hop-1 in Notch-pathway signaling. Proc. Natl. Acad. Sci. U. S. A. 96, 2497-2502. Abstract Article
Wilkin, M.B., Carbery, A.M., Fostier, M., Aslam, H., Mazaleyrat, S.L., Higgs, J., Myat, A., Evans, D.A., Cornell, M., and Baron, M. (2004). Regulation of Notch endosomal sorting and signaling by Drosophila Nedd4 family proteins. Curr. Biol. 14, 2237-2244. Abstract Article
Wilkinson, H.A., Fitzgerald, K., and Greenwald, I. (1994). Reciprocal changes in expression of the receptor lin-12 and its ligand lag-2 prior to commitment in a C. elegans cell fate decision. Cell 79, 1187-1198. Abstract Article
Wilson, J.J., and Kovall, R.A. (2006). Crystal structure of the CSL-Notch-Mastermind ternary complex bound to DNA. Cell 124, 985-996. Abstract Article
Wu, G., Hubbard, E.J., Kitajewski, J.K., and Greenwald, I. (1998). Evidence for functional and physical association between Caenorhabditis elegans SEL-10, a Cdc4p-related protein, and SEL-12 presenilin. Proc. Natl. Acad. Sci. U. S. A. 95, 15787-15791. Abstract Article
Wu, L., Aster, J.C., Blacklow, S.C., Lake, R., Artavanis-Tsakonas, S., and Griffin, J.D. (2000). MAML1, a human homologue of Drosophila Mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 26, 484-489. Abstract
Yochem, J., and Greenwald, I. (1989). glp-1 and lin-12, genes implicated in distinct cell-cell interactions in C. elegans, encode similar transmembrane proteins. Cell 58, 553-563. Abstract Article
Yochem, J., Weston, K., and Greenwald, I. (1988). The Caenorhabditis elegans lin-12 gene encodes a transmembrane protein with overall similarity to Drosophila Notch. Nature 335, 547-550. Abstract Article
Yoo, A.S., Bais, C., and Greenwald, I. (2004). Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulva development. Science 303, 663-666. Abstract Article
Yoo, A.S., and Greenwald, I. (2005). LIN-12/Notch activation leads to microRNA-mediated down-regulation of Vav in C. elegans. Science 310, 1330-1333. Abstract Article
*Edited by Jane E. Mendel. Last revised June 4, 2012. Published January 17, 2013. This chapter should be cited as: Greenwald I. and Kovall R., Notch signaling: genetics and structure (January 17, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.10.2, http://www.wormbook.org.
Copyright: © 2012 Iva Greenwald and Rhett Kovall. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: isg4@columbia.edu or kovallra@ucmail.uc.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.