Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
In C. elegans, mutants that are defective in muscle function and/or structure are easy to detect and analyze since: 1) body wall muscle is essential for locomotion, and 2) muscle structure can be assessed by multiple methods including polarized light, electron microscopy (EM), Green Fluorescent Protein (GFP) tagged proteins, and immunofluorescence microscopy. The overall structure of the sarcomere, the fundamental unit of contraction, is conserved from C. elegans to man, and the molecules involved in sarcomere assembly, maintenance, and regulation of muscle contraction are also largely conserved. This review reports the latest findings on the following topics: the transcriptional network that regulates muscle differentiation, identification/function/dynamics of muscle attachment site proteins, regulation of the assembly and maintenance of the sarcomere by chaperones and proteases, the role of muscle-specific giant protein kinases in sarcomere assembly and the regulation of contractile activity, and new insights into the functions of the dystrophin glycoprotein complex.
The nematode Caenorhabditis elegans has long been used to study muscle development, organization and function (Waterston, 1988; Moerman and Fire, 1997; the WormBook chapter Sarcomere assembly in C. elegans muscle). C. elegans has striated and non-striated muscles. Non-striated muscles include 20 pharyngeal muscle cells, 2 stomatointestinal muscles, one anal depressor muscle, one anal sphincter muscle, 8 vulval muscles, 8 uterine muscles, and 10 contractile gonadal sheath cells (http://www.wormatlas.org/hermaphrodite/musclenonstriated/mainframe.html). Most studies, however, have been performed on the 95 striated body wall muscle cells. These muscles are the functional equivalents of vertebrate skeletal muscles. The overall structure, composition, and function of the muscle basic functional unit, the sarcomere, is highly conserved between nematodes and vertebrates. Functional body wall muscle is required for the sinusoidal movement on semi-solid surfaces or the c-shaped thrashing of C. elegans in liquid. The optical transparency of the nematode allows visualization of muscle structure by polarized light microscopy, or fluorescence microscopy using GFP tagged proteins in live animals. Self-fertilization allows propagation of mutants that are unable to mate. Powerful forward and reverse genetics allows isolation and analysis of mutants in individual muscle components. All of these advantages have permitted research using C. elegans to make landmark discoveries for muscle in general, some of which are listed in Table 1, Section 8.
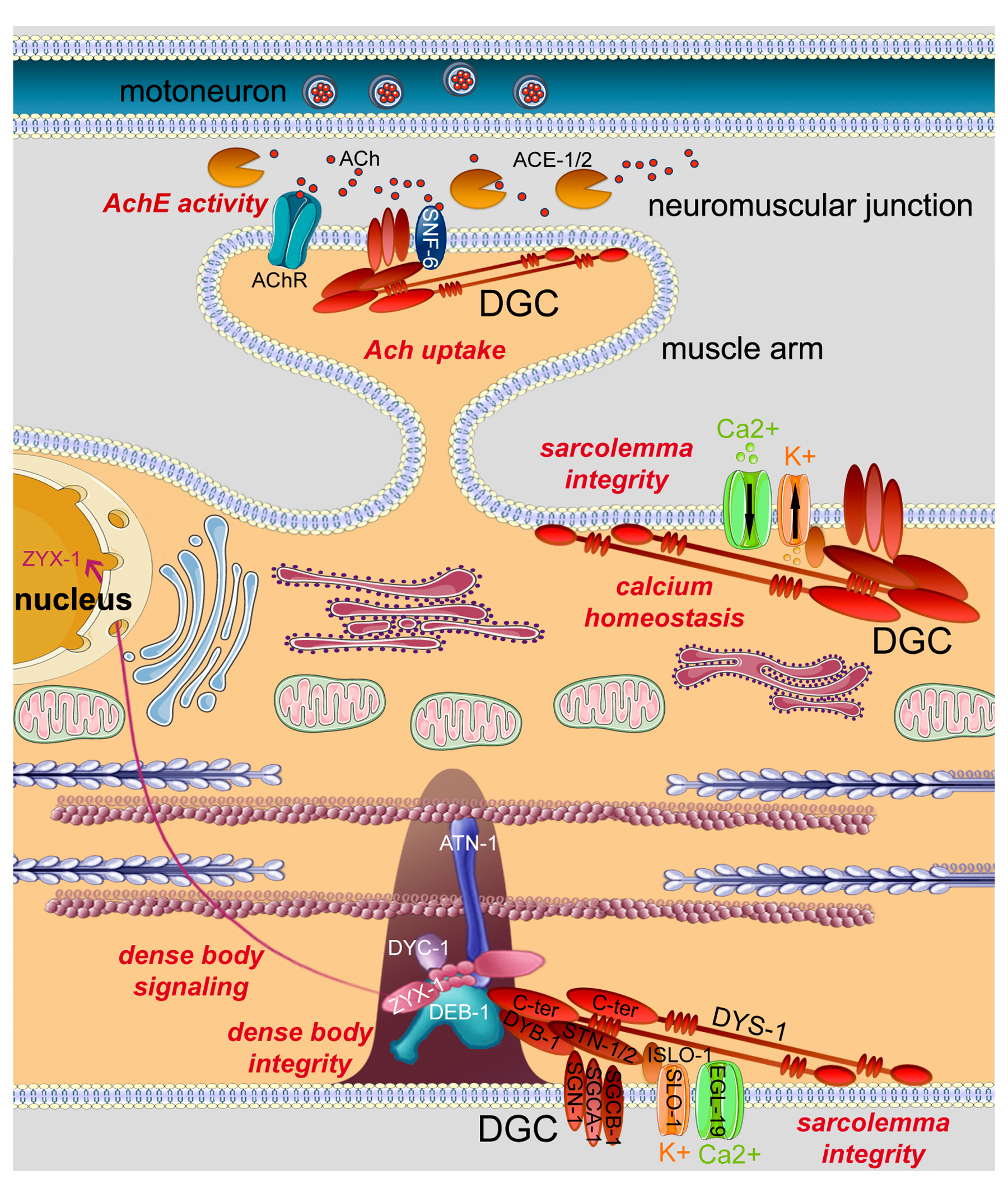
As in vertebrates, each sarcomere is composed of myosin containing thick filaments associated with an M-line, and actin containing thin filaments associated with a Z-disk analog named the dense body. The pulling of actin filaments by myosin heads generates force. To generate movement, the produced force needs to be transduced outside the muscle cell. In vertebrates, this force transduction is ensured by: 1) anchoring sarcomeres to the sarcolemma by specific muscle adhesion complexes, called costameres, that link Z-disks to the extra-cellular matrix (ECM) (Ervasti, 2003; Samarel, 2005); 2) by having all the myofibrils in a given cell mechanically linked by intermediate filaments; and 3) by the anchoring of the muscle ends through tendons to the bones. In C. elegans, M-lines and dense bodies directly perform the attachment of sarcomeres to the muscle membrane and the underlying basement membrane (Figure 1). These integrin based protein complexes thus share functional similarity with both the vertebrate Z-disk/ M-line and the costamere. In addition, the muscles are linked via fibrous organelles in the hypodermis to the underlying cuticle, the exoskeleton of the nematode (Cox and Hardin, 2004). Contractile filaments form a layer anchored to the distal sarcolemma of each cell (Figure 1). The striation observed by microscopy corresponds to the repetition of myosin-enriched A bands and actin-enriched I bands in alignment. While vertebrates exhibit cross-striated muscles, striation in C. elegans muscles appears slightly oblique with respect to the longitudinal axis of the muscle cell with which it forms an angle of 5.9° (MacKenzie and Epstein, 1980) (Figure 1).
|
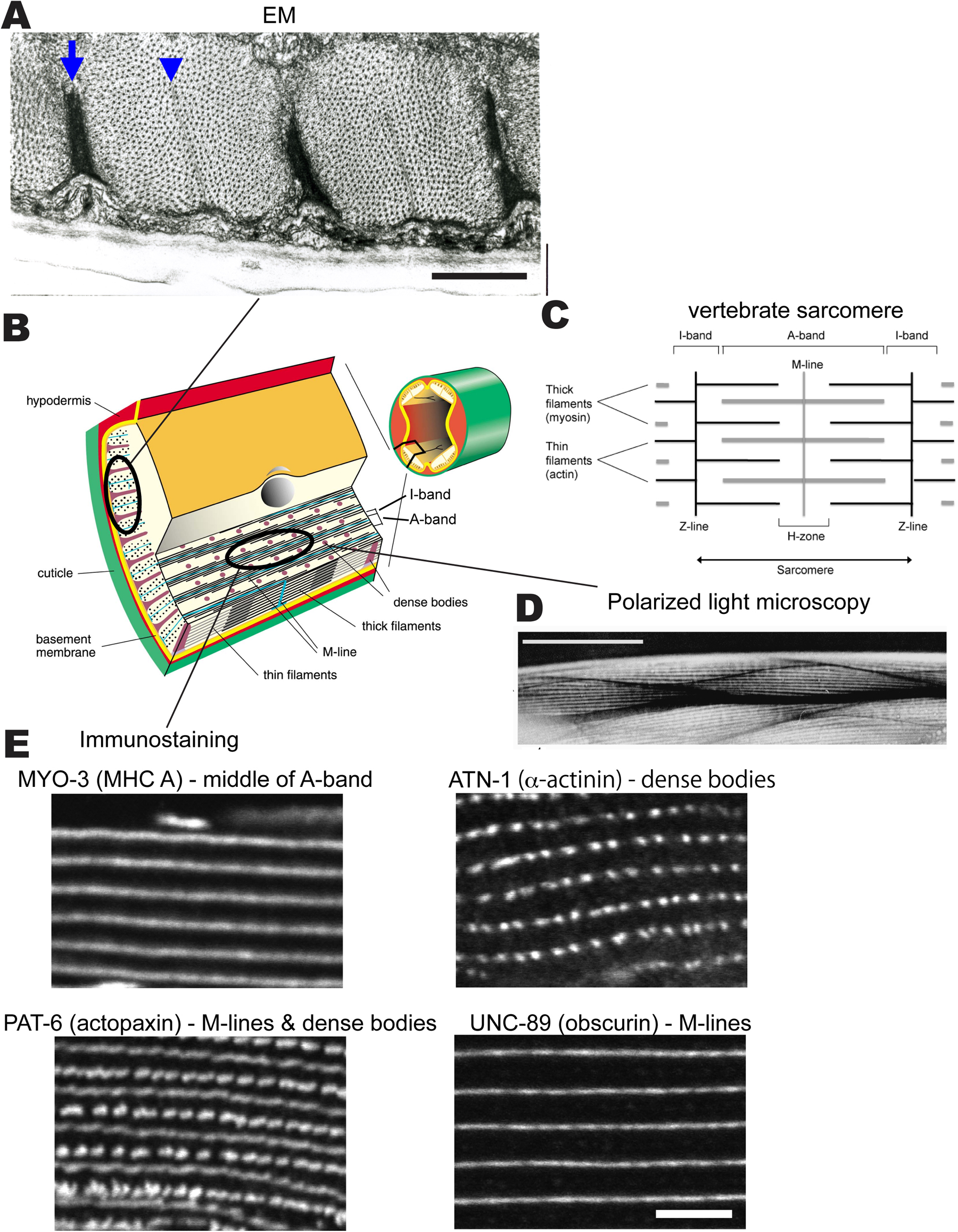
Figure 1. The body wall muscle of C. elegans. (A) Transmission electron microscopy (EM) of a cross-section of a body wall muscle cell showing two full sarcomeres. An arrow points to a dense body and an arrowhead points to an M-line. The largest black dots are cross sections of thick filaments in the A-bands; the smallest dots are cross sections of thin filaments in the I-bands (surrounding the dense bodies). Note that all the dense bodies and the M-lines are anchored to the muscle cell membrane, which sits on top of a basement membrane, a thin hypodermis, and thick cuticle. A diagonal line indicates how the EM view is related to the drawing in (B). Scale bar, 1 μm. (B) The small drawing on the right shows a cross-section through an adult worm emphasizing that the body wall muscle consists of four quadrants. Each quadrant consists of interlocking pairs of mononuclear spindle-shaped cells (23 or 24 per quadrant). In the enlargement, note that the myofilament lattice is limited to one side of the cell rather than filling the entire cross-sectional area as in a vertebrate striated muscle cell. Several planes of section are depicted, one of which emphasizes the muscle's striated organization with typical A-bands containing thick filaments organized around M-lines, and overlapping thin filaments probably attached to Z-disk-like structures called dense bodies. The sarcomere, which is defined as the repeating distance from one dense body to the next dense body is approximately 12 μm in adult muscle. Note that the plane of section parallel to the page is the plane viewed when an animal crawls on agar or is placed on a slide and examined by light microscopy (note the lines to the polarized light view in (D) and immunostaining views in (E). (C) A drawing of a typical sarcomere in vertebrate striated muscle. The sarcomere, which is defined as the repeating distance from one Z-line to the next Z-line is typically 2.2-2.5 μm. (D) Polarized light microscopy on a live nematode of portions of two muscle quadrants, each containing interlocking pairs of spindle shaped cells. The parallel white lines are A-bands, which alternate with parallel dark lines that are I-bands. Scale bar, 10 μm. (E) Immunofluorescent localizations of several sarcomeric proteins in adult muscle, MYO-3 (myosin heavy chain A, MHC A) localized to the middle of the A-bands; ATN-1 (α-actinin) localized to dense bodies; UNC-89 (obscurin) localized to M-lines; and PAT-6 (actopaxin) localized to both M-lines and dense bodies. Scale bar, 10 μm.
Most body wall muscle cells develop during embryogenesis. At hatching 81 muscle cells have formed; 14 muscle cells develop post-embryonically (Krause, 1995; Moerman and Fire, 1997). At the L1 larval stage muscle cells are 2 sarcomeres wide (Figure 2). During larval development new sarcomeres are added, resulting in adult cells that contain up to 10 sarcomeres in mid-body body wall muscle cells and smaller numbers of sarcomeres in body wall muscle cells at the ends of the animal (see WormBook chapter Sarcomere assembly in C. elegans muscle) (Figure 2). M-lines and dense bodies also increase in size during post-embryonic development. In adult muscles, the layer of the contractile apparatus, which is approximately 1.5 μm thick, is parallel to the hypodermis and the cuticle. Cellular organelles such as the nucleus, mitochondria, endoplasmic reticulum, ribosomes, etc. are localized in the deeper part of the muscle cell (Figure 1 and Figure 2).
|
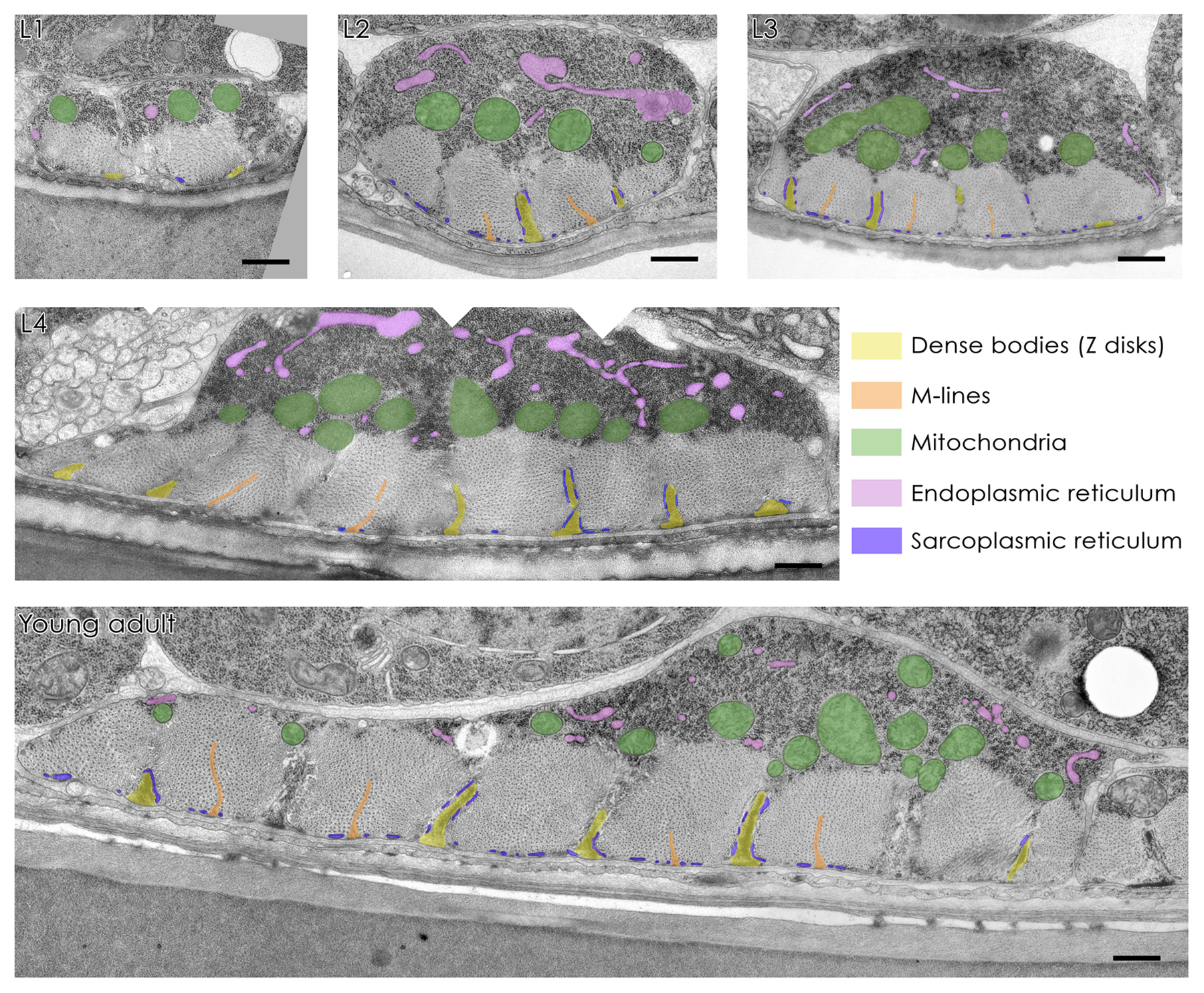
Figure 2. Muscle development observed by transmission EM. Electron micrographs are representative of muscle cells from wild type L1, L2, L3, and L4 larvae and young adults. All images were identically orientated with the cuticle at the bottom. Pseudo coloring indicates dense bodies (yellow), M-lines (orange), mitochondria (green), endoplasmic reticulum (pink), and sarcoplasmic reticulum (blue) localized around the dense bodies. Scale bars correspond to 0.5 μm in L1 to L4, and 0.2 μm in young adults. Figure adapted by N. Brouilly from Brouilly et al., 2015.
Some striking differences between nematodes and vertebrates make striated body wall muscles of adult C. elegans a tissue of choice to analyze cellular processes such as proteotoxicity, autophagy and mitophagy, mitochondrial biology, signaling pathways involving cell adhesion complexes, aging, and muscle degeneration. C. elegans muscles lack satellite cells (muscle stem cells) and therefore muscle regeneration. In adult worms, body wall muscles are totally post-mitotic, do not fuse and remain mononucleated. The adult worm possesses 95 diamond-shaped body wall muscle cells, which form a single layer of cells and are arranged in four longitudinal bands of two mutually offset rows of cells, named quadrants, running from head to tail. (http://wormatlas.org/hermaphrodite/musclesomatic/MusSomaticframeset/html). Together, these properties allow bypassing the complexities that arise from the syncytial nature of skeletal muscle of vertebrates.
Finally, it should be noted that, for practical reasons, we could not cover all of the interesting and important topics in nematode muscle. One major topic that is missing in our review is the role of actin regulatory proteins (e.g., UNC-60B (ADF/cofilin), UNC-78 (AIP1)) in sarcomere assembly and function. However, this subject has been expertly covered in a recent review by Ono (2014). We also do not discuss the effects of micro/zero gravity on muscle structure and function. Our review is primarily focused on sarcomere assembly and maintenance, rather than on the regulation of contractility. We do not fully discuss the role of K+ channels such as UNC-93/SUP-9/SUP-10, or BK channels, or Ca++ channels, although we do discuss the possible role of twitchin (UNC-22) in regulating contractility.
The 81 body wall muscle cells present in L1 larvae at hatching are derived from 4 of 5 founder blastomeres: one from AB, 28 from MS, 32 from C, and 20 from D. The germline founder cell, P4, does not produce any muscle cells. The D lineage produces exclusively body wall muscle cells, but other founder lineages produce multiple cell fates. Maternal factors that determine founder cell fate affect the number of body wall muscle cells. In skn-1 mutants, 28 body wall muscle cells that derived from MS are absent, but body wall muscle cells from C and D lineages are not affected (Bowerman et al., 1992; Bowerman et al., 1997). pal-1 mutants lack the body wall muscle cells from C and D lineages, but those from MS lineage are not affected (Hunter and Kenyon, 1996; Ahringer, 1997).
Embryonic development is controlled by maternally expressed genes initially, but then there is a switch to control by zygotically expressed genes (Baugh and Hunter, 2006). In the C and D lineages at least, pal-1 initially acts as a maternal factor for body wall muscle cell fate (Hunter and Kenyon, 1996; Edgar et al., 2001). pal-1 encodes a Caudal-related homeobox transcription factor (Hunter and Kenyon, 1996) and the maternal function of pal-1 is determination of C and D lineages; zygotically expressed pal-1 is directly involved in muscle differentation. Over-expression of pal-1 triggers robust muscle cell differentiation in the absence or reduced levels of POP-1 (TCF), the downstream transcriptional factor of Wnt/MAP kinase signaling. In the presence of high levels of POP-1 (TCF), over-expression of pal-1 produces robust hypodermal cells (Fukushige and Krause, 2005).
Myogenic regulatory factors (MRFs), first characterized in mammalian muscle, have also been characterized by genetic and molecular biological approaches in C. elegans (Moerman and Fire, 1997). hlh-1 encodes the C. elegans ortholog of MyoD and is the single MRF-related factor in C. elegans. Its alternative name, HLH-1, derives from having a basic helix-loop-helix domain (Krause et al., 1990). hlh-1 is expressed zygotically in embryonic body wall muscle precursor cells from about the 90 cell stage (Krause et al., 1990). hlh-1 null mutants stop development at the L1 larval stage with paralysis and severe morphological defects, suggesting essential roles of HLH-1 in development. However, in hlh-1 null mutant animals, the 81 embryonic body wall muscle cells are produced (Chen et al., 1992; Chen et al., 1994). These observations suggest that hlh-1 is important for development but not essential for myogenic differentiation, and that one or more other factors are involved in muscle differentiation. Ectopic expression experiments revealed that HLH-1 is a potent myogenic factor (Fukushige and Krause, 2005). When hlh-1 is ubiquitously expressed by a heat shock promoter in early embryos almost all cells adopt a muscle cell fate. Interestingly, although the cells that follow the muscle fate express many muscle markers, including structural attachment components (e.g., PAT-3 (β-integrin) and UNC-89 (obscurin)), there is no sarcomere assembly. The authors suggest that since the conversion to muscle fate abolishes other tissue fates, sarcomere assembly might require the presence of other tissues (e.g., the hypodermis) (Fukushige and Krause, 2005).
Over-expression of pal-1, which encodes maternal and zygotic factor functions in the C and D lineages, also converts embryonic cells to the muscle cell fate, and this conversion is not dependent on hlh-1, also suggesting that other factors independent from hlh-1 are involved in muscle differentiation (Fukushige and Krause, 2005). Microarray data suggest that two additional transcriptional factors, UNC-120 and HND-1, are involved in body wall muscle differentiation (Fukushige et al., 2006). First, unc-120 encodes a serum response factor (SRF)-related protein, and is induced by PAL-1 and HLH-1 (Dichoso et al., 2000; Baugh et al., 2005; Fukushige et al., 2006). Second, HND-1, which is closely related to mammalian HAND family basic helix-loop-helix proteins, is induced by PAL-1, but not by HLH-1 (Mathies et al. 2003; Fukuhsige et al., 2006). Ectopic expression of either unc-120 or hnd-1 also results in cells adopting the muscle cell fate (albeit less efficiently than when hlh-1 is ectopically expressed) including expression of muscle markers, suggesting that both transcriptional factors are also myogenic. Cells adopting a muscle cell fate occur by ectopic expression of HND-1 even in an hlh-1 null mutant background.
Muscle cell fates can also be induced by ectopic expression of UNC-120, and although this does not require hlh-1, the presence of hlh-1 increases the expression of muscle cell markers in these cells. An hlh-1, hnd-1, unc-120 triple mutant produces no body wall muscle cells in embryos, suggesting that these three transcriptional factors are required together for body wall muscle differentiation (Fukushige et al., 2006). Genetic analysis revealed that hlh-1 and hnd-1 are redundant, and that hlh-1 and unc-120 cooperate and maintain their expression until the late embryonic stage (Figure 3A). The PAL-1 protein directly binds to upstream enhancer regions of hlh-1 and unc-120 and activates their expression (Lei et al., 2009). The binding of HLH-1 to an upstream enhancer region of the hlh-1 gene makes the positive auto-regulation of hlh-1 possible (Krause et al., 1994) (Figure 3B). Target genes for HLH-1 have been identified; 2,753 by ChIP-seq, and 1,032 by ChIP-Chip (Lei et al., 2010). An overlapping set of 569 target genes was identified by both methods. Future analysis of these genes will hopefully clarify the transcriptional network that is regulated by HLH-1. It is interesting to note that Meissner et al. (2009) identified a total of 3,577 genes expressed in body wall muscle by SAGE (Serial Analysis of Gene Expression) and microarray data. Among the 2,753 genes identified as targets of HLH-1 by ChIP-seq, 880 genes are found in the Meissner set; among the 1,032 genes identified as targets of HLH-1 by ChIP-chip, 282 are found in the Meissner set. Among the 569 genes identified by both ChIP methods, 194 are also found in the 3,577 genes identified by Meissner et al. (2009).
|
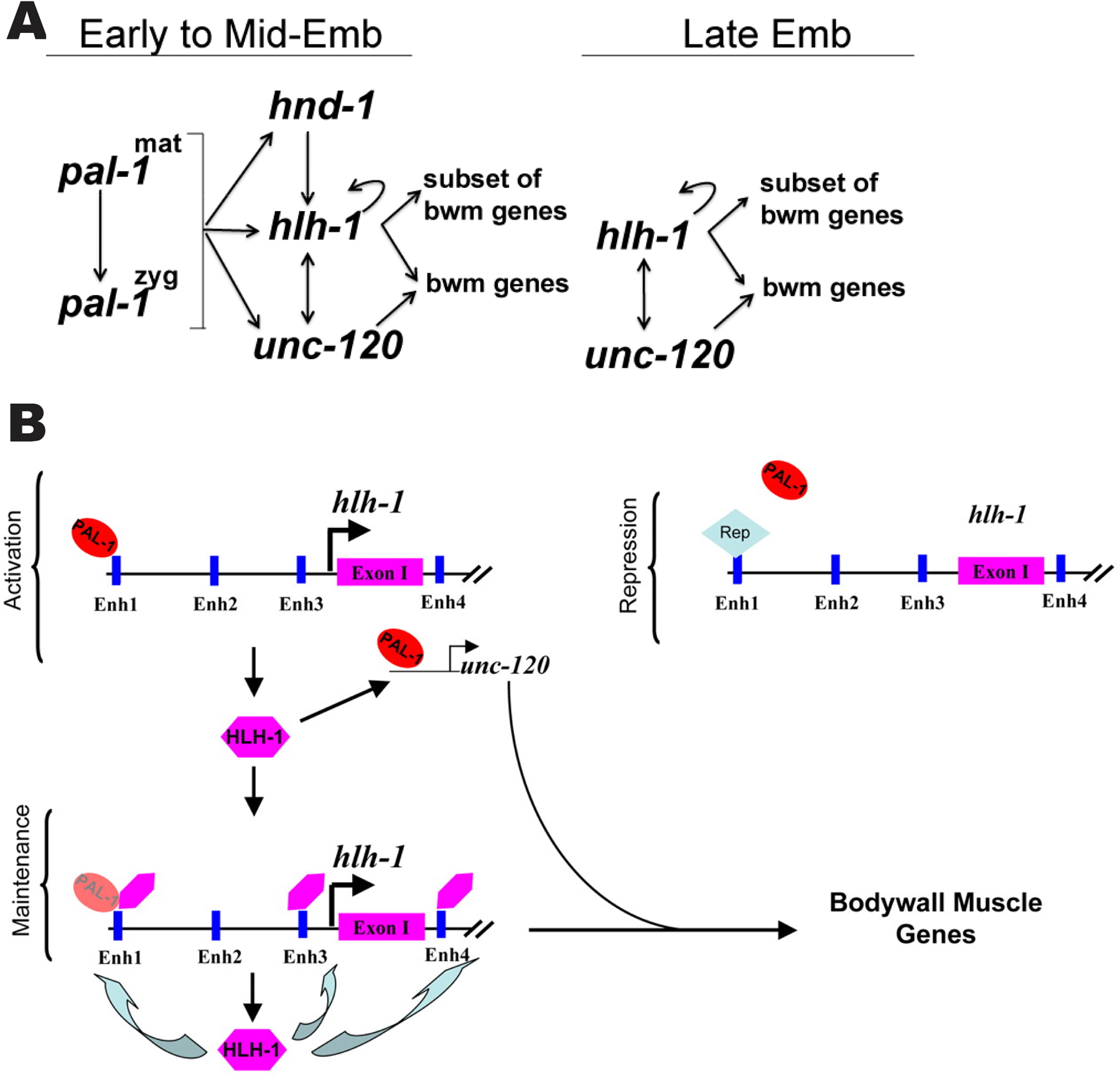
Figure 3. (A) Transcriptional regulation of body wall muscle cells in the C and D founder cell lineages of C. elegans embryos. Arrows indicate genetic relationships. Maternal and zygotic PAL-1 activates expression of HLH-1, UNC-120, and HND-1. All three transcription factors are involved in cell fate determination of body wall muscle. HLH-1 and UNC-120 continue to express until late embryonic stage. (Adapted, with permission, from Fukushige et al., 2006). (B) A model for activation of muscle-expressing genes by PAL-1. PAL-1 binds to enhancer regions of hlh-1 and possibly unc-120. HLH-1 binds to multiple enhancer regions of hlh-1 itself, resulting in positive auto-regulation. HLH-1 also binds to an enhancer region of unc-120 and activates its expression. (Adapted, with permission, from Lei et al., 2009).
In addition to being expressed in embryos, hlh-1 continues expression in mature body wall muscle cells including in adults (Krause et al., 1994; our unpublished results). This suggests that HLH-1 has a role in promoting the continued development of larval and adult muscle, and perhaps even maintenance of adult muscle.
In vertebrate muscle differentiation, MRFs cooperate with other transcription factors, such as MEF-2 and Twist. In C. elegans, the MEF-2 ortholog is not essential for myogenesis (Dichoso et al., 2000). However, it should be noted that MEF-2, like UNC-120, is related to SRF and it could be that nematodes became reliant on UNC-120 (SRF), whereas other animals became reliant on MEF-2. The C. elegans Twist homolog, HLH-8, is expressed in mesodermal tissues (M cell lineage) (Harfe et al., 1998). hlh-8 null mutants have egg-laying and defecation defects. Corresponding to this phenotype, in hlh-8 null mutants sex muscle and defecation muscles are not formed properly: vulval muscles are missing, and 5% of the animals have fewer, and 60% have extra sex myoblasts. In hlh-8 nulls, although the overall organization of body wall muscle is normal, the number of postembryonic body wall muscle cells is different: 15% of the animals have extra body wall muscle cells (96-100 cells), and 80% have fewer body wall muscle cells (86-94 cells) (Corsi et al., 2000).
The first success in identifying muscle attachment components came from an immunological approach. Francis and Waterston (Francis and Waterston, 1985; Francis and Waterston, 1991) used protein fractions enriched in nematode body wall muscle components to generate a battery of monoclonal antibodies and then determined their staining patterns in nematodes and their Western blot reactivity. These monoclonal antibodies recognized many components of muscle and hypodermis attachment structures, and the extracellular matrix.
Identification of the genes involved began with the identification of myo-3, the gene encoding body wall muscle myosin heavy chain A (MHC A) (Waterston, 1989). Body wall muscle contains two myosin heavy chain isoforms, MHC A and MHC B (Epstein et al., 1974; Miller et al., 1983). These isoforms form homodimers (Schachat et al., 1978) and are differentially localized in the 10 μm long adult thick filaments, with MHC A lying in the central 2 μm, and MHC B lying in the polar 4 μm of the thick filament (Miller et al. 1983). unc-54, which encodes myosin heavy chain B (MHC B), was first identified genetically (Epstein et al., 1974) while the other 3 myosin heavy chain genes expressed in muscle, including myo-3, were first identified through sequence homology (Miller et al., 1986). unc-54 null mutant animals show reduced movement at all stages and as adults are unable to move on an agar surface. Although duplication and two-fold overexpression of myo-3, was identified as a suppressor (called sup-3) of unc-54 null animals (Riddle and Brenner, 1978; Maruyama et al., 1989), the loss-of-function phenotype for myo-3 was not known. The two genes, unc-54 and myo-3 are of equivalent size, and while many alleles of unc-54 had been identified, none had been identified for myo-3. This led Bob Waterston to wonder if myo-3 loss-of-function (lof) might be lethal. He designed a screen for lethal mutations linked to sma-1, as myo-3 had been localized close to this gene by in situ hybridization (Albertson, 1985). He obtained multiple lethal mutants that arrested at different developmental stages, two of which bore similarity to certain unc-54 dominant alleles when homozygous. These two candidate mutants failed to complement and sequencing revealed point mutations in the myo-3 gene (Waterston, 1989). The myo-3 mutant embryos stopped elongation at the 2-fold stage and showed greatly reduced or no movement in the eggshell although other aspects of development including pharyngeal pumping and cuticle deposition were normal; the embryos hatched, remained in a folded state, did not move, and died.
Soon after the discovery of the myo-3 phenotype, it was shown that strong loss-of-function alleles for the unc-45 gene display a similar embryonic lethal phenotype (Venolia and Waterston, 1990). Using two of the aforementioned antibodies developed by Francis and Waterston (1985), which recognize proteins localized to the base of dense bodies, Barstead and Waterston (1989) screened an expression library, and pulled out cDNAs specifying C. elegans vinculin, encoded by the deb-1 gene. After placing deb-1 on the genetic map, a screen was conducted for loss-of-function mutations (Barstead and Waterston, 1991). Two mutants in deb-1 were shown to be embryonic lethal and displayed the same phenotype that was first identified for loss-of-function mutations in the myo-3 gene (Waterston, 1989).
The phenotype of loss-of-function alleles consistent with a null state for myo-3, unc-45 and deb-1 is “Pat”, which is an abbreviation for paralyzed and arrested at two-fold embryonic stage. During normal nematode embryonic development there is both an expansion of cell number and morphogenesis in which the initially football-shaped embryo elongates 4-fold, going through stages that are named according to length, 1.5-fold, 2-fold, and 3-fold, before hatching from the eggshell. At the ≈1.5-fold stage, embryonic muscle contraction begins and the embryos continue to move within the eggshell throughout the remainder of embryogenesis. Pat embryos do not move in the eggshell at the 1.5-fold stage (or later) and arrest development at the 2-fold stage. Other aspects of development continue and the embryo hatches as an abnormal “jack knife”-appearing L1 larva and dies.
Encouraged by findings of Pat phenotypes for myo-3, deb-1, and unc-45, Williams and Waterston (1994) conducted a genome-wide screen and identified 13 additional genes with a Pat mutant phenotype, and new alleles of deb-1 and myo-3. Three classes of Pat mutants were isolated, based on severity of paralysis in the eggshell: severe Pat mutants were similar to the original myo-3 and deb-1 mutants; “Mild Pat” mutants (pat-11, pat-12), in which movement began at the 1.5 fold stage but did not show vigorous movement at the 2 fold stage; and “Lat” mutants (late paralysis, arrested elongation at two fold) (let-2, emb-9), in which movement began at the 1.5 fold stage and continued vigorously, as in wild type, through the 2 fold stage. The Pat genes were placed into five classes (I to V in lessening severity), based on the localization and organization of myosin and actin. Class I genes (unc-52, unc-112, pat-2, pat-3) showed no localization of myosin or actin near the sarcolemma (lack of “polarization”). Class II genes (pat-4, pat-6, pat-11) showed polarization but no organization of myosin or actin into thick or thin filaments. Class III genes (deb-1, pat-8, pat-9, pat-12) showed both normal polarization and thick filaments but no thin filaments. Class IV genes (lev-11, pat-5, pat-10) showed normal polarization and normal thick and thin filaments. Class V consisted of one gene, myo-3, and showed normal polarization, normal thin filaments, but no thick filaments. At the time this study was completed, a number of the gene products had been identified: unc-52 encodes the ECM protein perlecan (Rogalski et al., 1993), pat-3 encodes β-integrin (Gettner et al., 1995), and deb-1 encodes vinculin (Barstead and Waterston, 1989). In addition, there was unpublished data cited noting that pat-2 was likely to encode β-integrin (Gettner et al., 1995; see also WormBase pat-2 page). Therefore, the authors proposed that because class I genes have the most severe effects and encode components of the ECM and sarcolemma, sarcomere assembly proceeds from the outside to the inside, initiated by molecules localized at the cell surface at future dense bodies and M-lines. This excellent model was verified by studying the temporal appearance of adhesion and sarcomere components during embryonic muscle development (Hresko et al., 1994), and by the subsequent cloning and mutant analysis of the other Pat genes, primarily by the laboratories of Don Moerman and Ben Williams (summarized in Sarcomere assembly in C. elegans muscle).
The other major phenotypic class of muscle-affecting mutant genes is the “Unc” (uncoordinated) class of 40 genes. Mutations in any of these genes result in slow moving or paralyzed adult worms (Waterston et al., 1980; Zengel and Epstein, 1980). There are a total of 111 Unc genes, but 71 of these genes primarily affect the nervous system, not muscle. For a number of muscle unc genes (unc-45, unc-52, unc-97, and unc-112), the phenotype of hypomorphic alleles is Unc, and the phenotype of null alleles is Pat. unc-112 encodes the nematode ortholog of mammalian Kindlins (Rogalski et al., 2000; Qadota et al., 2014), pat-4 encodes integrin linked kinase, ILK (Mackinnon et al., 2002), and pat-6 encodes the nematode ortholog of actopaxin (Lin et al., 2003). unc-97 encodes the C. elegans ortholog of PINCH (Hobert et al., 1999), and the null state for unc-97 is also Pat (Norman et al., 2007). All the above-mentioned pat and unc gene products, except for unc-45, have been localized to both dense bodies and M-lines by GFP fusions (Hobert et al., 1999; Rogalski et al., 2000; Mackinnon et al., 2002: Lin et al., 2003), and also in some cases by specific antibodies (Gettner et al., 1995; Mullen et al., 1999; Lin et al., 2003; Hikita et al., 2005; Miller et al., 2006; Qadota et al., 2012; Warner et al., 2013).
Yeast two hybrid (Y2H) assays and binding experiments using purified proteins have demonstrated that class I and II Pat gene products interact with each other (Figure 4). Based on what is known about their mammalian orthologs, it is likely that UNC-52 (perlecan) associates with PAT-2 (β-integrin) and PAT-3 (β-integrin) at the extracellular surface of muscle cells (Figure 5). Inside the muscle cell, the cytoplasmic tail of PAT-3 associates directly with UNC-112 (Qadota et al., 2012); PAT-4 associates with UNC-112 (Mackinnon et al., 2002), with PAT-6 (Lin et al., 2003), and with UNC-97 (Mackinnon et al., 2002; Norman et al., 2007). The UNC-112/PAT-4/PAT-6/UNC-97 complex has been confirmed by co-immunoprecitation (Qadota et al., 2014). UNC-52, PAT-2, PAT-3, UNC-112, PAT-4, PAT-6, and UNC-97 are found at the base of both M-lines and dense bodies.
|
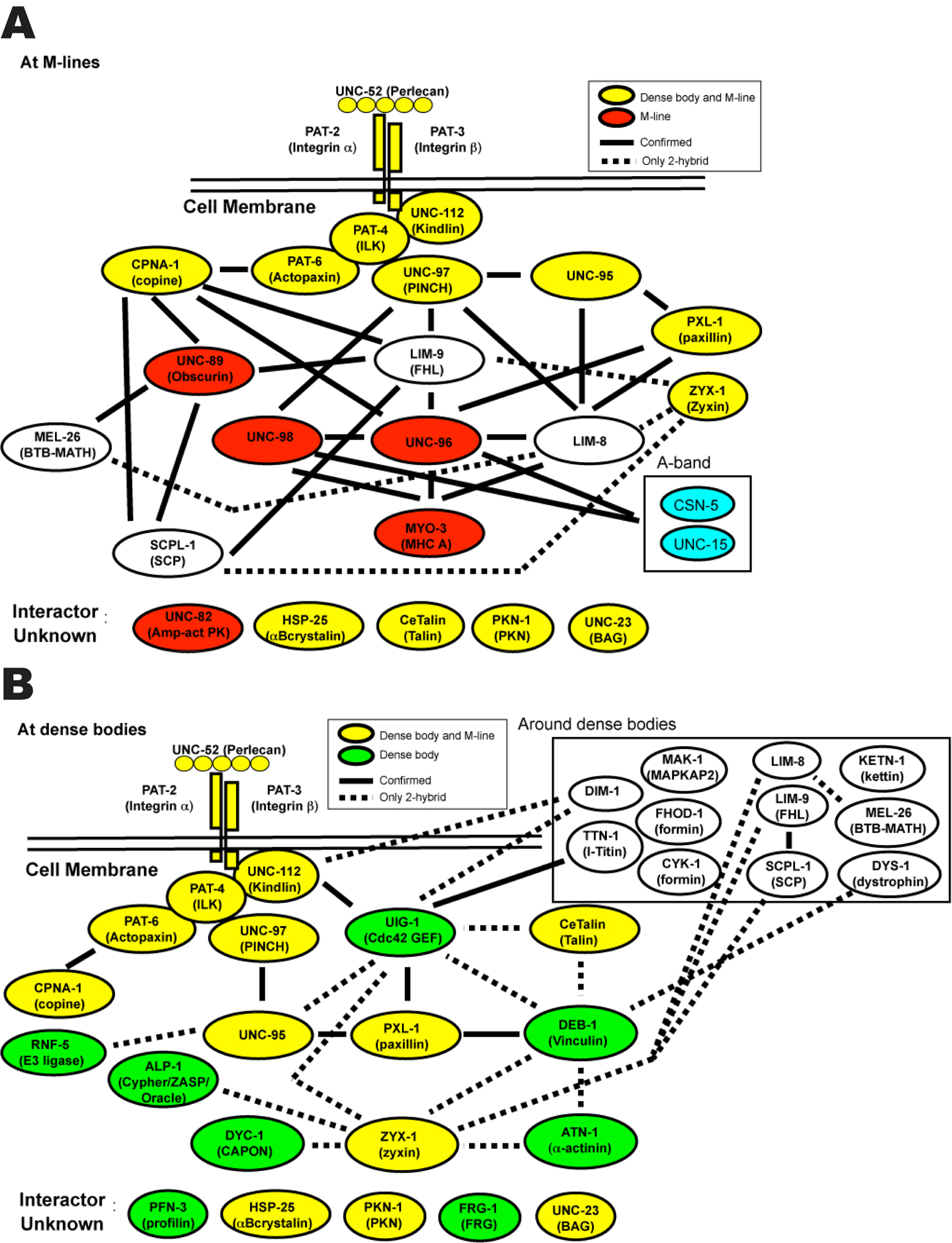
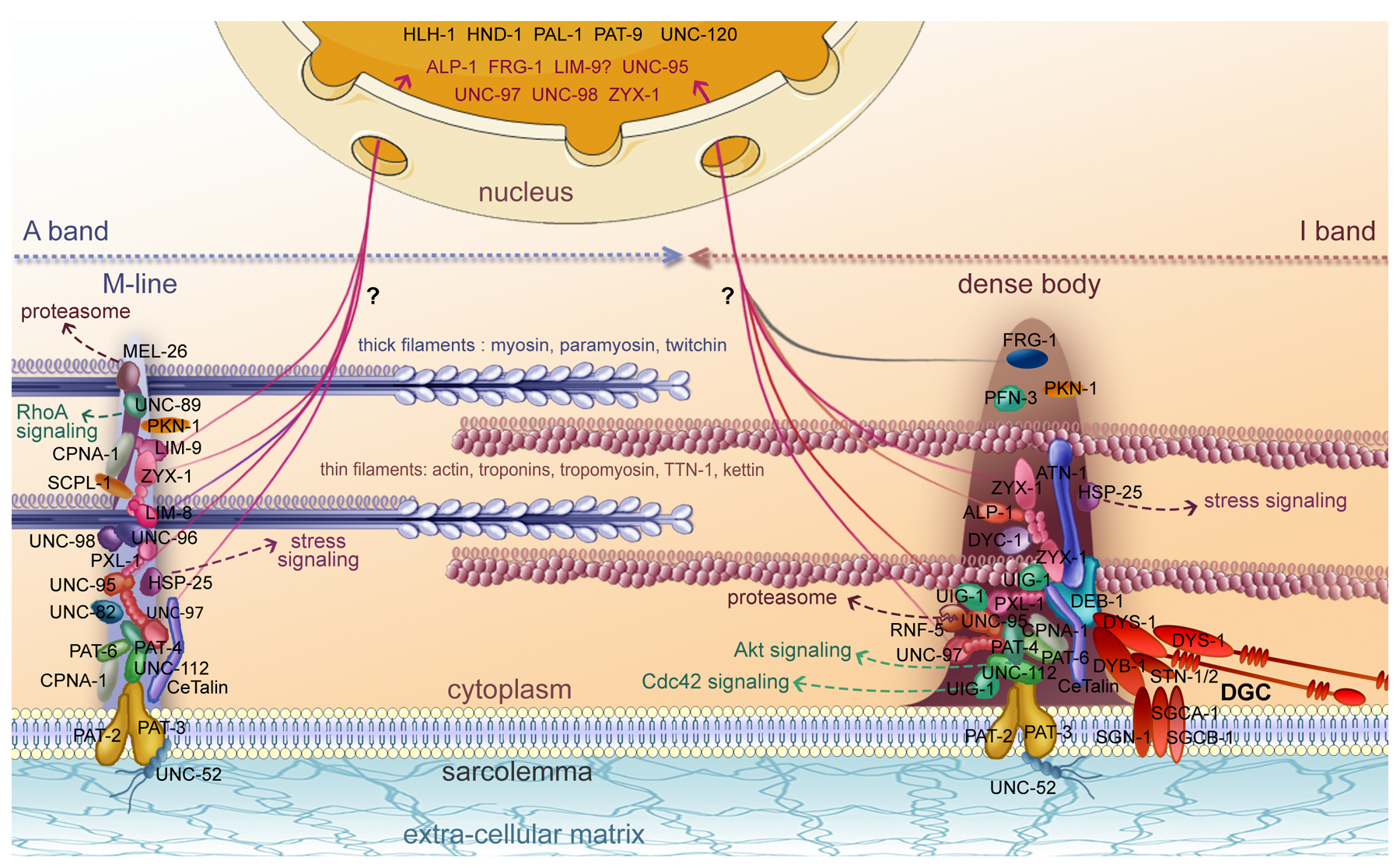
Figure 4. Networks of interacting proteins found at integrin adhesion sites in body wall muscle of C. elegans. Yellow denotes proteins that are localized to both M-lines and dense bodies; red denotes proteins that are localized only to M-lines; green denotes proteins localized only to dense bodies; white (or clear) denotes proteins that are found around or between dense bodies, and in some cases, also to M-lines; blue indicates proteins that are localized to the A-band. In parentheses are the names of the human orthologs or homologs. Most of the interactions were first identified by Y2H screens or assays, and then confirmed by in vitro binding using purified recombinant proteins, localized by antibodies and/or GFP fusions, and genetic analysis. Black lines indicate confirmed interactions; dotted lines indicated interactions only suggested by Y2H results. At M-lines, these interactions create a physical linkage from the muscle cell membrane (via integrins) to MHC A in thick filaments. At dense bodies, beginning also with integrins, there is a similar linkage of multiple proteins, presumably to actin in thin filaments, via DEB-1 and ATN-1. A few of the interactions (MEL-26 to LIM-8; UNC-95 to LIM-8; CeTalin (TLN-1) to UIG-1; CeTalin to DEB-1; DEB-1 to ATN-1; UIG-1 to TTN-1; UNC-112 to DIM-1; UIG-1 to DIM-1) are unpublished (H. Qadota, D.G. Moerman, G.M. Benian, unpublished data).
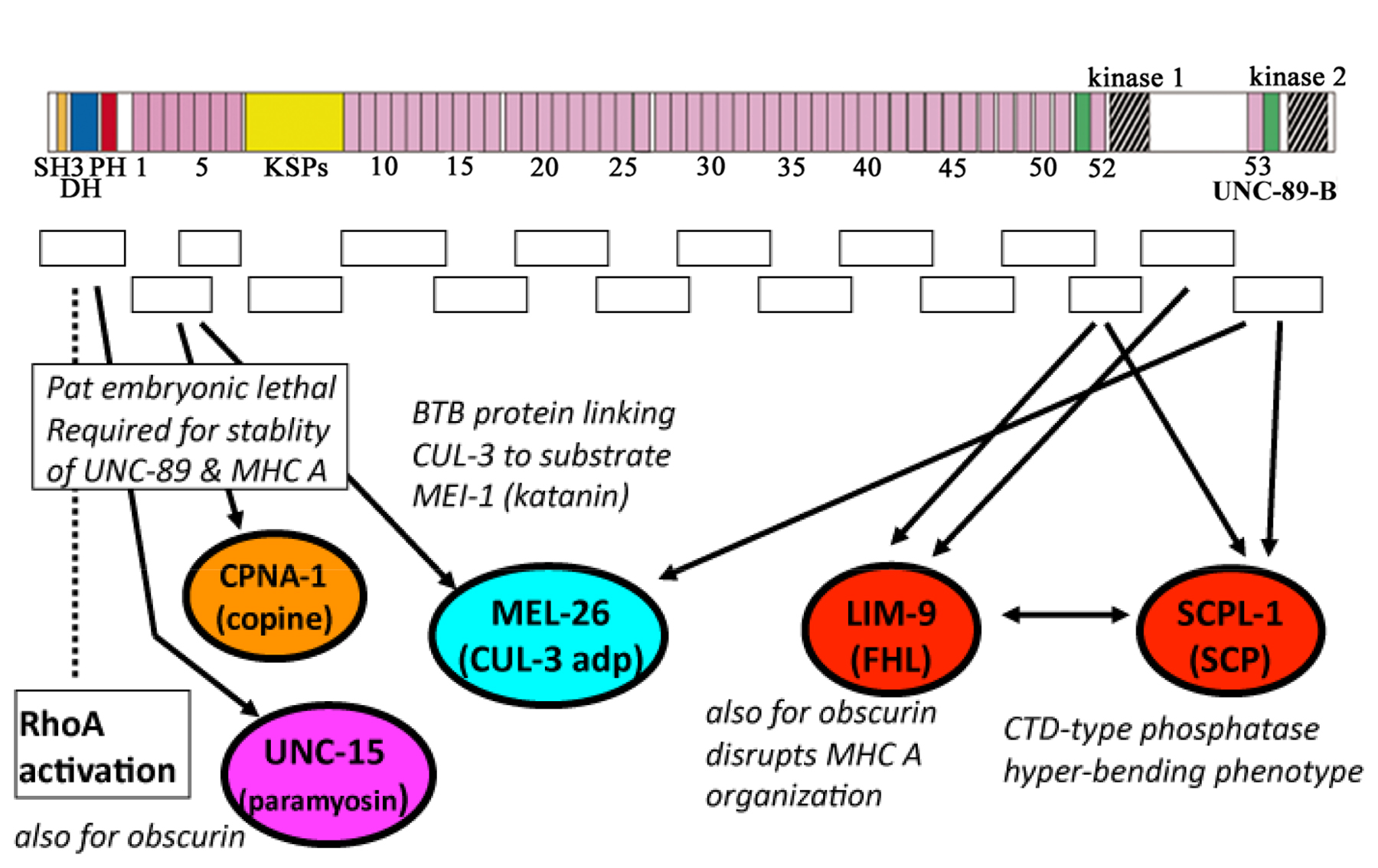
Three additional proteins have been localized at both M-lines and dense bodies: ZYX-1 (zyxin) (Lecroisey et al., 2008; Lecroisey et al., 2013; discussed below), HSP-25 (αβ-crystallin) (Ding and Candido, 2000), and CeTalin (Moulder et al., 1996). CeTalin is now called TLN-1, and is also known as UNC-35 (WormBase). Although the effect of unc-35 mutations on body wall muscle structure and function have not been reported, the fact that at least one allele (e259) (Brenner, 1974) leads to an Unc phenotype indicates the importance of this protein in muscle. Some of the M-line-specific proteins (UNC-89, UNC-98, UNC-96, UNC-82) are described below.
In addition to DEB-1 (vinculin) (Barstead and Waterston, 1989; discussed above), dense-body specific proteins include: ATN-1 (α-actinin) (Barstead et al., 1991), UIG-1 (Cdc42 GEF) (Hikita et al., 2005), and ALP-1 (ALP/Enigma) (McKeown et al., 2006). ATN-1 was identified by homology to vertebrate α-actinin and its gene placed on the genetic map (Barstead et al., 1991); it is the sole α-actinin gene in C. elegans. An atn-1 null mutant (Moulder et al., 2010) shows abnormally short and broad dense bodies by EM, but most of the I-bands are normally ordered and, as shown by phalloidin staining, display some abnormal accumulations of F-actin near the sarcolemma. Despite these abnormalities, the atn-1 null mutant has a remarkably mild locomotion defect: it shows normal locomotion in liquid (swimming/thrashing) but has less ability to bend maximally, consistent with a defect in the transmission of muscle contraction (discussed below). UIG-1 was identified as a binding partner for UNC-112 from a Y2H screen (Hikita et al., 2005). UIG-1 shows Cdc42-specific guanine nucleotide exchange activity in vitro. GFP::UIG-1 localizes to dense bodies; a uig-1 intragenic deletion mutant shows abnormal adult muscle structure by polarized light. Figure 4 and Figure 5 represent a more complete picture of proteins residing at both M-lines and dense bodies, or specifically at one of the structures. Nevertheless, this is a tentative picture that is likely simplified, based on what is known for vertebrate adhesion complexes (Zaidel-Bar and Geiger, 2010). Some details about the proteins shown in Figure 5 and discussed in the text, including human homologs/orthologs, location in the sarcomere, interacting partners, phenotypes of mutants, and key references are given in Table 2, Section 8.
|
Figure 5. Molecular composition of the sarcomere in body wall muscles. The cartoon shows a schematic representation of part of a sarcomere. Thin filaments (pink) are mainly composed of actin (ACT-1 to 4), tropomyosin (LEV-11), troponin C (PAT-10), troponin I (TNT-1 to 3), and troponin T (MUP-2). Kettin (KETN-1), and TTN-1 are likely associated to thin filaments. Thick filaments (blue) contain myosin heavy chains A (MYO-3) and B (UNC-54), paramyosin (UNC-15), and twitchin (UNC-22). Thick filaments are likely cross-linked at the M-line, and thin filaments are probably anchored to the dense body. The main proteins associated to dense bodies and M-lines are indicated along with some of the protein interactions summarized in Figure 4. The bases of dense bodies and M-lines are composed of α- (PAT-2) and β- (PAT-3) integrin dimers (yellow). Integrin anchors dense bodies and M-lines to the sarcolemma and likely interact with perlecan (UNC-52) at the outside of muscle cells. Some dense body and M-line associated proteins are linked to signaling pathways: UNC-89 to RhoA signaling, UIG-1 to Cdc42 signaling, HSP-25 to stress signaling, UNC-112 to Akt signaling, and MEL-26 and RNF-5 to the ubiquitin proteasome system. ALP-1, FRG-1, UNC-95, UNC-97, UNC-98, ZYX-1, and most likely LIM-9, possibly shuttle from the dense body and/or the M-line into the nucleus. Several transcriptional regulators are permanently present in the nucleus: HLH-1, HND-1, PAL-1, PAT-9, and UNC-120. Dystrophin (DYS-1) and the Dystrophin Glycoprotein Complex (DGC, red) localize close to the dense body. By the interaction of its C-terminal end with DEB-1, DYS-1 may link the DGC to the dense body. The DGC is composed of dystrobrevin (DYB-1), δ-and β-sarcoglycans (SGCA-1 and SGCB-1), δ/γ-sarcoglycan (SGN-1) and syntrophins (STN-1 and -2). This figure was adapted by C. Lecroisey from Lecroisey, 2010.
Localization of UNC-112 to muscle integrin adhesions requires PAT-4 (Mackinnon et al., 2002). PAT-4 (ILK) kinase domain binds to the N-terminal half of UNC-112 (Mackinnon et al., 2002). UNC-112 N- and C-terminal halves interact, and UNC-112, but not PAT-4, interacts directly with the cytoplasmic tail of PAT-3 (Qadota et al., 2012). Biochemical, genetic, and cell biological data (Qadota et al., 2012; Qadota et al., 2014) suggest the following model for this requirement: UNC-112 exists in a closed form that interacts weakly with integrin, and an open form that interacts more strongly with integrin, and conversion to the open active form is promoted by binding of PAT-4 (ILK) to UNC-112 (Figure 6A).
|
Figure 6. Conformational model for the interaction of UNC-112 with integrin, and key mutations supporting the model. (A) It is hypothesized that UNC-112 exists in two conformations, closed and open, and that only the open form can interact with the cytoplasmic tail of β-integrin at the base of adhesion sites in muscle. The conversion to the open active form is promoted by binding of UNC-112 to PAT-4. (B) UNC-112 D382V does not bind to PAT-4, and fails to localize to integrin adhesion sites in vivo. According to the model, this mutant form of UNC-112 is in a closed conformation. (C) In wild type UNC-112, the N- and C-terminal halves interact. However, UNC-112 T346A or UNC-112 E349K fails to show this interaction. UNC-112 containing both D382V and T346A (or D382V and E349K) does localize to adhesion sites in vivo. According to the model, this is because even without PAT-4 binding this mutant form of UNC-112 is in an open conformation able to bind to integrin. (D) PAT-4 P257L allows PAT-4 to bind to UNC-112 D382V. Co-expression of UNC-112 D382V and PAT-4 P257L allows for localization of UNC-112 D382V to adhesion sites in vivo. (E) Schematic representation of domains, mutation sites and interaction of UNC-112 with PAT-4. Domains of each protein, indicated by colored rectangles were predicted by PFAM, with boundaries indicated by residue numbers. The black and purple brackets denote the minimal regions that are required for interaction of UNC-112 with PAT-4, respectively. In UNC-112, the D382V and E302G mutations each prevent UNC-112 from interacting with PAT-4; the T346A and the E349K mutations each prevent interaction of the N-terminal half of UNC-112 with the C-terminal half of UNC-112, and in the presence of D382V or E302G, act as intragenic suppressors to permit binding of these mutant forms of UNC-112 to PAT-4. In PAT-4, P257L is one of 9 mutations lying within the pseudo kinase domain that act as extragenic suppressors of UNC-112 D382V, allowing this mutant form of UNC-112 to interact with PAT-4. Underlined residues are conserved in human kindlins and ILK. The original genetic mutations in unc-112 and pat-4 are not shown. (Updated, with permission, from Qadota et al. 2012; Qadota and Benian, 2014).
To obtain evidence for this model in vivo, UNC-112 and PAT-4 non-binding mutant forms and their suppressor mutations were isolated (Figure 6E). This was accomplished by error-prone PCR mutagenesis followed by Y2H screening, and confirmation using in vitro binding assays with recombinant proteins. UNC-112 D382V and UNC-112 E302G cannot bind to PAT-4 (Figure 6E). When UNC-112 D382V is expressed in worms, it does not localize to integrin adhesion sites in striated muscle, suggesting that PAT-4 binding is required for UNC-112 localization to these sites (Figure 6B). Using the same mutagenesis/Y2H methods, two classes of suppressor mutations (Figure 6E) for D382V were isolated. One class consists of intragenic suppressors in UNC-112: T346A and E349K mutations block the interaction of UNC-112 N- and C-terminal halves. UNC-112 (D382V and T346A (or E349K)) cannot bind to PAT-4. However, UNC-112 (D382V and T346A (or E349K)) can localize to integrin adhesion sites in worms, suggesting that a change of UNC-112 to an “open” conformation is essential for association with integrin (Figure 6C). The second class consists of extragenic suppressors, and these are missense mutations in PAT-4. Each of the nine PAT-4 suppressor mutant proteins can bind to UNC-112 D382V (one of nine is P257L) (Figure 6E). In the presence of PAT-4 P257L, UNC-112 D382V can localize to integrin adhesion sites in vivo (Figure 6D). Using the crystal structure of human ILK (Fukuda et al., 2009), an homology model of PAT-4 was generated, and this shows that all nine mutations affect residues that may form a binding surface for UNC-112, and lie on a different surface from that which binds to α-parvin (PAT-6). Confirmation of this model awaits a crystal structure of UNC-112 and/or the UNC-112/PAT-4 complex. Currently, however, a crystal structure of any kindlin (like UNC-112) is not available.
As is apparent in Figure 4 and Figure 5, the same proteins are located at the base of both dense bodies and M-lines, and these are the earliest proteins to associate in the assembly process, as shown by immunostaining during embryonic muscle development (Hresko et al., 1994). However, a mystery is what determines whether a dense body or an M-line will be built upon this same “foundation”? Certainly, this involves the recruitment of dense body-specific and M-line-specific proteins, but it is not known how this occurs. Dense body-specific proteins include DEB-1 (vinculin) (Barstead and Waterston, 1989; discussed above), ATN-1 (α-actinin) (Barstead et al., 1991), UIG-1 (Cdc42 GEF) (Hikita et al., 2005), and ALP-1 (ALP/Enigma) (McKeown et al., 2006; discussed below). M-line specific proteins include UNC-89, UNC-98, UNC-96, and UNC-82 (discussed below). One way to approach this problem might be to conduct screens for mutants in which a GFP-tagged protein that is normally present only at the dense body or the M-line, is found at both structures, or the opposite structure.
It should be noted that the various components of M-lines and dense bodies are not uniformly distributed throughout these structures. At both M-lines and dense bodies, because UNC-112 interacts with the cytoplasmic tail of PAT-3 (β-integrin), UNC-112 and its associated proteins (PAT-4, UNC-97, and PAT-6) are located close to the outer muscle cell membrane. For example, a confocal z-series shows that UNC-52 (perlecan) and PAT-6 (actopaxin), via antibodies, and UNC-112::GFP, are located close to the cell membrane (Warner et al., 2013). This also seems to be true for UNC-95, which interacts with UNC-97 (Qadota et al., 2007), and DIM-1 (Ig domain protein ) (Rogalski et al., 2003). At dense bodies, DEB-1 (vinculin) is clearly near the muscle cell membrane whereas ATN-1 (α-actinin) is clearly located in the major and deeper region of dense bodies (Francis and Waterston, 1985). The ATN-1 (α-actinin) binding protein, ALP-1, is localized to the deeper portion of dense bodies, similar to ATN-1 (Han and Beckerle, 2009). Interestingly, ZYX-1 is located in the middle of dense bodies, consistent with its interaction with both DEB-1 (vinculin) and ATN-1 (α-actinin) as demonstrated by Y2H assays (Lecroisey et al., 2013). By a z-series of confocal images, UNC-89 (Warner et al., 2013) is located throughout the depth of the M-line, from near the outer muscle cell membrane to deep into the myofilament lattice. CPNA-1 is found at all three levels of dense bodies (membrane proximal, middle, and deep), but at M-lines, CPNA-1 is found near the muscle cell membrane, absent from the middle, and then reappears deep in the lattice. However, this may reflect either absence in the middle portion of the M-line, or that CPNA-1 epitopes are masked by other proteins in this region (Warner et al., 2013).
There are also a number of proteins that are found around and between dense bodies (as indicated in Figure 4). These include LIM-8, LIM-9 (FHL-2) (Qadota et al., 2007), the formin family members FHOD-1 and CYK-1 (Mi-Mi et al., 2012), MEL-26 (Wilson et al., 2012), DIM-1 (Rogalski et al., 2003), and MAK-1 (MAPKAP kinase 2) (Matsunaga et al., 2015). TTN-1 (Forbes et al., 2010; see below) and KETN-1 (kettin, an invertebrate-specific actin-binding 427,000 Da protein comprised of 31 Ig domains) (Ono et al., 2006) are more evenly distributed throughout the I-band (except for dense bodies).
GFP tagged proteins can be used to determine the real-time dynamic exchange of proteins in vivo. The first, and so far, most extensive study on the dynamics of sarcomeric proteins in C. elegans has been reported by Ghosh and Hope (2010). The authors used Fluorescence Recovery After Photobleaching (FRAP) to assess the mobility of six GFP tagged proteins, UNC-112 (kindlin), UNC-95, MYO-3 (MHC A), MUP-2 (troponin T), T03G6.3 (NPP6), and C46G7.2 (large isoform of α-filagenin), in living transgenic young adult worms. After photobleaching, recovery of fluorescence is interpreted as replacement of bleached proteins with proteins that arrive from either a cytoplasmic pool, or are coming off from similar structures outside the bleached area; given the timescale of the typical experiment contribution from newly synthesized protein is negligible. The proteins studied showed a wide variation of exchange rates. Thin filament or I-band components, MUP-2 (troponin T) and T03G6.3 (NPP6) were fastest to recover, with t1/2 of recovery of 2.5 min and 1.5 min, respectively. MYO-3 (MHC A), C46G7.2, and UNC-112 (kindlin) showed no recovery even after 15 min. UNC-95 showed an intermediate response, with about 50% recovering initially at a fast rate comparable to T03G6.3, and then maintaining that level up through 15 min (Ghosh and Hope, 2010). As noted by the authors, their results for the replacement of MUP-2 and MYO-3 are consistent with what has been reported for thin filament and thick filament components in vertebrate and zebrafish skeletal muscles; rapid exchange for thin filament components, slow exchange for myosin. The authors suggest that the more dynamic proteins are peripheral components of complexes or filaments, whereas the less dynamic proteins are more central proteins with roles in anchorage. Their results with the M-line/dense body protein UNC-112, is certainly consistent with this interpretation (Ghosh and Hope, 2010).
More recently, the dynamics of ZYX-1 (zyxin) have been studied in living zyx-1::gfp transgenic worms (Lecroisey et al., 2013). FRAP experiments indicate that ZYX-1 is highly dynamic. When a region containing dense body and M-line ZYX-1::GFP was bleached, the average t1/2 of recovery was 4.96 sec, with maximal recovery of 77.5% occurring after 70 sec. When nuclei were bleached, the average t1/2 of recovery was 7.57 sec, with a maximum recovery of only 14.3% after 70 sec. The lower maximum recovery for nuclear ZYX-1::GFP suggests that a large percent (~86%) of ZYX-1 may be permanently localized to the nucleus. In contrast, the faster maximum recovery for muscle attachment complex ZYX-1 suggests that ZYX-1 is more peripheral and dynamic in these locations. Preliminary FRAP experiments on UNC-98::GFP have also been performed (R.K. Miller and G.M. Benian, unpublished data). After photobleaching nuclear UNC-98::GFP, the t1/2 of recovery was 163 sec; after photobleaching a line of 4 dense bodies, the t1/2 of recovery was 25 sec. However, after photobleaching the M-line, the fluorescence did not even reach half of its original level during the 725 sec monitoring period. These results suggest that nuclear UNC-98 may be more stable than nuclear ZYX-1, and that although UNC-98 is very stably associated with the M-line, it is only peripherally or transiently associated with the dense body (see comment above that endogenous UNC-98, may be localized to the M-line, not the dense body).
The fast recovery rates of the nematode integrin adhesion complex proteins ZYX-1, dense body-associated UNC-98, and UNC-95 is consistent with FRAP results obtained for integrin, tensin, talin, and ILK at the myotendinous junctions of Drosophila embryonic and larval muscles: each of these proteins had t1/2 of less than 100 sec (Yuan et al., 2009). Overall, FRAP experiments in C. elegans and Drosophila demonstrate that integrin adhesion structures show considerable turnover of their components. This turnover may play an important role in the maintenance of these structures, which are subjected to mechanical stress during muscle activity.
unc-95, unc-96, and unc-98 were first identified by Zengel and Epstein (1980) from a screen for mutants that are defective in muscle function and structure. unc-96 and unc-98 mutants are slower moving than wild type, and by polarized light microscopy display a moderately disorganized myofilament lattice and birefringent “needle-like” structures at the ends of their body wall muscle cells. These “needles” correspond to accumulations of proteins that contain paramyosin, but not actin, myosin, UNC-89, or α-actinin (Mercer et al., 2003; Mercer et al., 2006; Miller et al., 2008). UNC-98 is a 310-residue polypeptide containing four C2H2 Zn finger domains and several predicted nuclear localization and nuclear export signal sequences (NLS and NES) (Mercer et al., 2003). Antibodies to UNC-98 localize to M-lines. However, in transgenic animals, UNC-98::GFP localizes to M-lines, dense bodies, and muscle cell nuclei. unc-98 mutant animals, when rescued with a wild type copy of the gene, show localization of anti-UNC-98 antibodies to M-lines, dense bodies, and nuclei. Deletion derivatives of UNC-98::GFP in transgenic worms demonstrated that the N-terminal 110 residues of UNC-98 are necessary and sufficient for nuclear localization, and that all four Zn fingers are sufficient for localization to M-lines and dense bodies (Mercer et al., 2003). Using an UNC-98 bait to screen a collection of Y2H clones representing known M-line and dense body proteins, interaction with UNC-97 (PINCH) was identified and confirmed by in vitro binding using purified proteins. Binding requires the first two LIM domains of the five LIM-domain protein UNC-97, and all four C2H2 Zn fingers of UNC-98 (Mercer et al., 2003).
unc-96 encodes 408 and 418 residue polypeptides by alternative splicing, and these proteins lack recognizable domains (Mercer et al., 2006). Antibodies to UNC-96 localize to M-lines (Mercer et al., 2006). The strongest mutant allele of unc-96, sf18, is not Pat embryonic lethal, and yet is presumably a null mutant as it is a nonsense mutation and no protein can be detected by Western blot. Intriguingly, either a decreased (by loss-of-function mutation) or an increased level (by over-expression from a heat shock promoter) of UNC-96 results in disorganization of thick filaments (Qadota et al., 2007). Apparently, the level of UNC-96 must be tightly controlled to obtain proper organization of thick filaments. By both genetic and biochemical criteria, UNC-96 and UNC-98 interact with each other. Protein accumulations at the ends of the muscle cells contain the UNC-98 protein in unc-96 mutants, and contain the UNC-96 protein in unc-98 mutants (Mercer et al., 2006).
unc-95 was identified at the molecular level by Broday et al. (2004). unc-95 mutants are slow moving and have disorganized muscle structure. Immunostaining with various antibodies shows that thick and thin filaments and dense bodies are disorganized (Broday et al. 2004). UNC-95 is a 350-residue polypeptide with a single LIM domain near its C-terminus, a region predicted to have coiled-coil structure and a NLS sequence (Broday et al., 2004). In transgenic nematodes, UNC-95-GFP localizes to M-lines, dense bodies, muscle cell-cell boundaries, and nuclei in adult body wall muscle. UNC-95::GFP is also expressed in embryonic muscle, and by the 3-fold stage is localized to muscle attachment sites (dense bodies and M-lines) and nuclei. Antibodies to UNC-95 clearly label M-lines, dense bodies and cell-cell boundaries, but not nuclei in adult body wall muscle (Qadota et al., 2007).
Genetic, cellular, and biochemical evidence support a model in which UNC-98 links integrin-associated proteins to myosin in thick filaments at M-lines (Miller et al., 2006) (Figure 4A). As noted above, UNC-97 (PINCH), a member of a conserved four-protein complex associated with integrin, interacts with UNC-98 (Mercer et al., 2003). The N-terminal 110 residues of UNC-98 interact with the C-terminal portion of myosin heavy chain A (MHC A) that resides in the middle of thick filaments in the proximity of M-lines (Miller et al., 2006). Although vertebrate costameres are usually regarded to reside at the level of Z-disks, some components of focal adhesions, including αv integrin have also been found at M-lines (McDonald et al., 1995). Thus, these data for C. elegans muscles suggest the possibility of a similar mechanism of linkage between integrins and myosin thick filaments at the M-lines of peripheral myofibrils of vertebrate muscle. UNC-97, in addition to interacting with UNC-98, also interacts with LIM-8, LIM-9, and UNC-95 (Qadota et al., 2007). These proteins are involved in three additional linkages from UNC-97 to myosin: 1) from UNC-97 to LIM-8 to myosin, 2) from UNC-97 to LIM-9 to UNC-96 to myosin, and 3) from UNC-97 to UNC-95 to LIM-8 to myosin. LIM-8 is a novel LIM domain-containing protein. LIM-9 is the nematode homolog of mammalian FHL-2 (four and a half LIM domain protein 2). UNC-96 and LIM-8 also bind to the C-terminal portion of MHC A (to a slightly different portion of MHC A that binds to UNC-98) (Figure 4A). These interactions were first identified by Y2H and then confirmed by in vitro binding assays using purified proteins. By antibody staining, LIM-8, LIM-9, and UNC-95 localize, at least partially, to M-lines. The fact that UNC-96, UNC-98, and LIM-8 interact with C-terminal portions of the myosin rod is consistent with models for the M-line in which the shafts of thick filaments are cross-linked.
In addition to a structural role for UNC-96 and UNC-98 at the M-line, these proteins interact with paramyosin (UNC-15) to promote paramyosin's incorporation into thick filaments (Mercer et al., 2006; Miller et al., 2008). Paramyosin is an invertebrate-specific “headless myosin” that is primarily an α-helical coiled-coil rod and is 36-38% identical in sequence to the rod domains of myosin heavy chains (Kagawa et al., 1989). In C. elegans body wall muscle, the myosins and a portion of paramyosin are organized around a tubular core consisting of paramyosin and filagenins in a specific geometry (Deitiker and Epstein, 1993; Epstein et al., 1995; Muller et al., 2001). In C. elegans, paramyosin is encoded by a single gene, unc-15. Loss-of-function unc-15 mutants are severely paralyzed and display disorganized body wall muscle (Waterston et al., 1977). unc-15 null mutants have shorter hollow-appearing thick filaments (MacKenzie and Epstein, 1980).
The birefringent needles observed in the body wall muscle of unc-96 and unc-98 mutants contain paramyosin located outside the thick filaments (Mercer et al., 2006; Miller et al., 2008). By genetic and biochemical criteria, paramyosin interacts with UNC-96 and UNC-98 (Mercer et al., 2006; Miller et al., 2008). By both Y2H analysis and ELISAs using purified proteins, UNC-98 interacts with paramyosin residues 31-693, whereas UNC-96 interacts with a separate region of paramyosin, residues 699-798. Although UNC-96 and UNC-98 affect, at least partially, the localization of paramyosin (some in accumulations, some in its normal A-band location), total paramyosin levels do not change in either unc-96 or unc-98 loss-of-function mutants (Miller et al., 2008). By Western blot, in the unc-15 nonsense mutant e1214, the level of UNC-98 is diminished, and in unc-15 missense mutants (e1215 and e73, which form paramyosin aggregates), the level of UNC-98 is increased. The dependence of UNC-98, and possibly UNC-96, levels on the state of paramyosin might be due to a chaperone function of these proteins in response to aggregation of mutant paramyosin. In fact, there is growing evidence that molecular chaperones are crucial for muscle assembly and maintenance; this topic is discussed in detail in Section 4.
There are two more links between paramyosin and M-line proteins. First, is the connection of paramyosin to UNC-82. unc-82 mutants have disorganized sarcomeres including mis-localization of thick filament and M-line components (Hoppe et al., 2010). Phenotypic analysis suggests that unc-82 is required in embryonic muscle cells, as the cells, including the myofilaments, increase in size. In an unc-82 null mutant, at the 1.5 fold stage of embryogenesis, the localization of MHC A, paramyosin, and UNC-89 are normal, but by the 2 fold stage, these proteins form aggregates, which become larger and more numerous at the 3 fold stage. Muscle contraction per se does not seem to be responsible for aggregate formation, as an unc-54(s95);unc-82 double mutant, in which myosin activity is reduced, does not fully rescue the phenotype. UNC-82 is a 1600 residue polypeptide containing an N-terminal protein kinase domain homologous to human ARK5 (NUAK1) and SNARK (NUAK2) protein kinases, and simple repetitive sequences in the remainder of the protein. A rescuing UNC-82::GFP fusion protein localizes to M-lines throughout the depth of the myofilament lattice. UNC-82 is a candidate for a protein kinase that phosphorylates myosin heavy chains and paramyosin (Hoppe et al., 2010). A small N-terminal nonhelical segment of paramyosin is phosphorylated on serine by a thick filament-associated kinase (Dey et al., 1992; Schriefer and Waterston, 1989). This N-terminus contains multiple copies of the motif, S_S_A, which may be sites of phosphorylation. This S_S_A motif is also found in multiple copies in the C-terminal nonhelical tailpieces of MHC A and MHC B. Supporting the idea that UNC-82 phosphorylates paramyosin is the observation that more acidic isoelectric species of paramyosin are absent from extracts of unc-82 mutants (Schriefer and Waterston, 1989).
Another link between paramyosin and M-line proteins is that the M-line protein UNC-89 interacts with paramyosin through its SH3 domain, and unc-89 mutants that lack expression of UNC-89 isoforms containing the SH3 domain show aggregates of paramyosin (Qadota et al., 2016; see below).
Null mutants for many of the M-line and dense body proteins do not have defects in sarcomere organization, or even locomotion when observed by a conventional motility assay in which the number of times a worm moves back and forth (flexes) in liquid is counted (as described in Epstein and Thomson, 1974). One possibility for this lack of effect could be functional redundancy. The functional redundancy of PXL-1 and LIM-8 (described in more details below) provides an example: neither loss-of-function mutant by itself has a defect in body wall muscle structure or nematode locomotion, however, a pxl-1; lim-8(RNAi) animal is slow moving with disorganized sarcomeres (Warner et al., 2011). Another possibility is that some of these proteins may not have roles in muscle assembly or maintenance. Instead, the M-line, and especially the dense body, is a “way station” for proteins to “park” or “rest” before departing for a new location. This is a tempting idea, but there is no evidence for it currently.
Yet another possibility is that these proteins, located as they are at muscle focal adhesions, function in force transmission, but a more sensitive assay is needed to reveal this function (Qadota and Benian, 2010). The ability to maximally bend during backward movement (Figure 7A) can discriminate wild type from many of the mutants in various integrin adhesion site proteins. The first successful application of this assay was on a null mutant of the single α-actinin gene in the worm, atn-1. Homozygous null mutations in this gene yield animals with abnormally short and broad dense bodies, but surprisingly display normal movement on an agar surface and normal swimming in liquid (Moulder et al., 2010). In contrast, the more sensitive bending assay shows that this mutant has a reduced ability to bend (Moulder et al., 2010). When the assay was applied to mutants in 18 other genes encoding proteins that are located at M-lines and dense bodies, defective bending was found in 12 more (Nahabedian et al., 2012). Four of them, unc-82, unc-89, unc-95, and unc-96 were previously described as having reduced motility. Thus, this assay can detect a locomotion defect in mutants that were previously recognized to have an adult “Unc” phenotype. Eight of the mutants were not previously known to have motility defects (pkn-1, zyx-1, frg-1, alp-1, kin-32, pfn-3, lim-8, and dim-1). Interestingly, one mutant gene, scpl-1 (2 alleles tested), had greater ability to bend maximally. SCLP-1 is a CTD-type phosphatase found to interact with UNC-89 (Qadota et al., 2008a; see below).
|
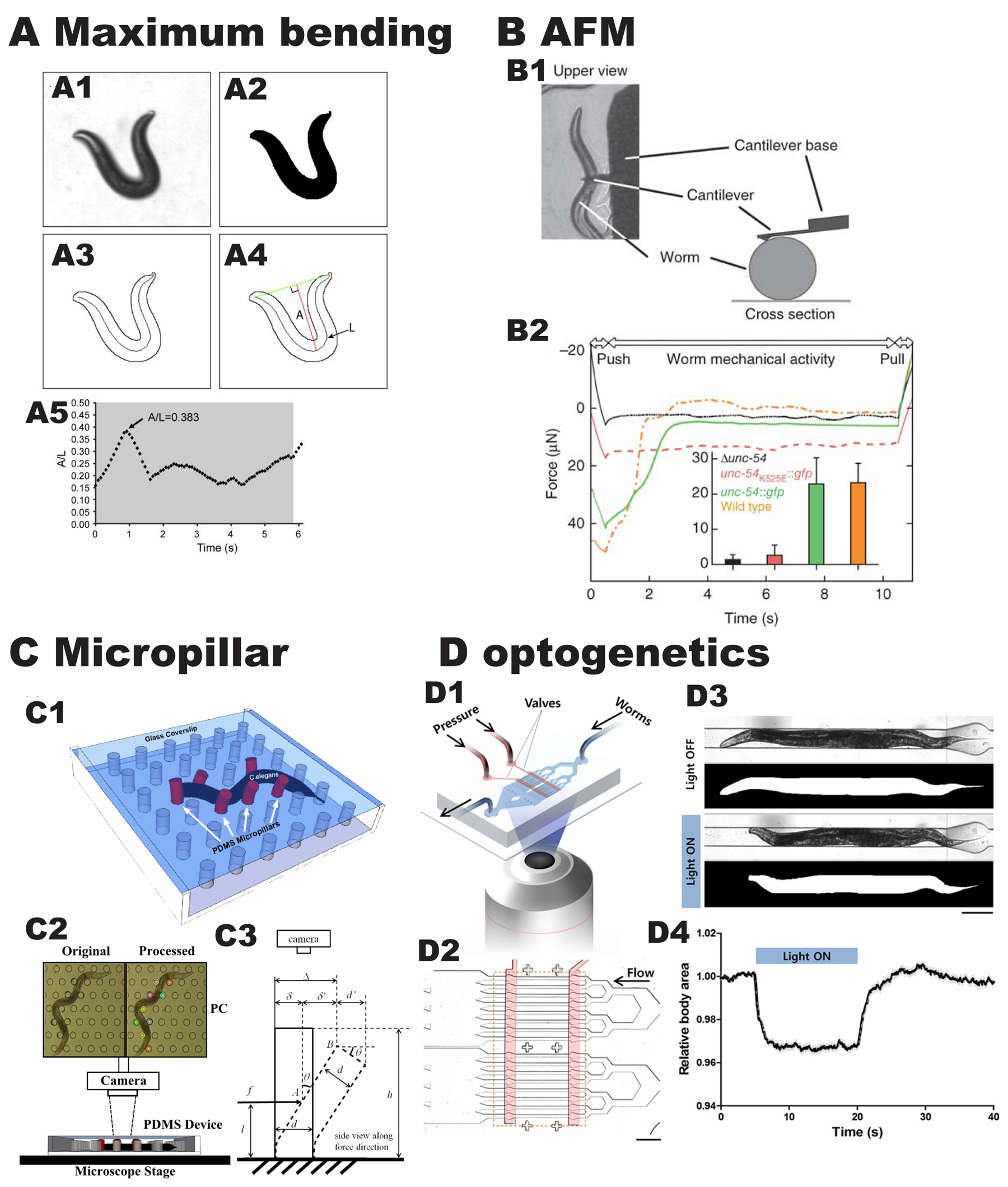
Figure 7. Recently reported methods to assess the locomotion and force generated by C. elegans. (A) Measurement of maximum bending amplitude (A/L) as a worm moves backwards. (A1) Frame from a video showing a deep bend of an adult during a reversal. (A2) Binary image of the frame from (A1). (A3) Outline of the animal and the corresponding midline spline used to determine the length of the animal. (A4) Measurement of the amplitude of the bend was made by measuring the longest perpendicular length, A, from a line connecting the head to the tail into the midline spline, of coutour length L. The ratio of this value, A, to the contour length, L, was calculated (A/L). (A4) Time course of A/L values. Shortly before t = 0, the worm was tapped gently on the head to induce backwards movement. The grey region indicates the time the animal is in a reversal. Reproduced from Nahabedian et al. (2012) with permission. (B) The use of an atomic force microscope (AFM) to measure force generated by a single nematode. (B1) Schematic of how the experiment is setup. (B2) Representative force curves generated by four C. elegans strains. Animals were exposed to a deforming force (push), and their force-generating activities were measured (worm mechanical activity) until the retraction of the cantilever (pull). The inset shows the force production averages and error bars with standard deviations of 15-25 animals from each strain. Reproduced with permission from Varkuti et al. 2012. (C) Measurement of the force generated by single nematodes by measuring deflection of flexible micropillars. (C1) Drawing of an animal as it deflects an array of micropillars made from polydimethylsiloxane (PDMS). (C2) Experimental setup comprising the PDMS device on a microscope stage with a camera connected to a computer for recording worm videos. (C3) Schematic of the deflected pillar for C. elegans force measurement model. Reproduced with permission from Johari et al. (2013). (D) Use of optogenetics to measure the kinetics of muscle contraction and relaxation. (D1) Experimental setup that combines microfluidics, optogenetics and image processing. (D2) The microfluidic device has 16 parallel microchannels for simultaneous illumination and analysis of multiple animals trapped by pneumatically-controlled microvalves (red). (D3) Microscopic and segmented images of a wild-type worm trapped in a microchannel with (bottom) and without (top) the illumination of blue light. (D4) Temporal change in the body size of wild type animals that was calculated from the segmented images and normalized by average value in the first 5 seconds (s). The light was on for 15 s beginning at the 5 s time point. Data represent mean +/- standard error of the mean, n = 88. Reproduced with permission from Hwang et al. (2016).
Varkuti et al. (2012) reported a new and conserved actin binding site on the myosin head called the “activation loop” that is crucial for F-actin activation of myosin ATPase activity. In this same report, the authors describe a novel method for measuring worm motility, and a novel method for measuring the force exerted by single nematodes (Figure 7B). After verifying that a missense mutation in this activation loop affects myosin's biochemical properties in vitro (with Dicytostelium and mouse myosins), the comparable mutation K525E was introduced into UNC-54 (MHC B), and a transgenic strain was created in which P unc-54::UNC-54K525E::GFP was introduced into an unc-54 null background. These animals showed reduced locomotion as compared to wild type or transgenic animals in which the unc-54 null was rescued with wild type UNC-54::GFP. Locomotion was measured by calculating velocities of worm tails in video recordings. To measure the ability of individual worms to generate force a clever method was devised in which a single worm was placed beneath the cantilever of an atomic force microscope (AFM), worms were exposed to a deforming force (a “push”), and the force the worm generates to escape from under the cantilever was measured. Force (μNewtons) vs. time (sec) show that wild type and the unc-54 null mutant rescued by UNC-54::GFP generated ≈30-50 μN, whereas UNC-54K525E::GFP generated much less force (~15-18 μN). It would be informative to use this method to measure forces generated by other alleles of unc-54 and many other Uncs as well.
Johari et al. (2013) used another innovative method to measure the force generated by individual wild-type nematodes (Figure 7C). This assay records the movement of a worm in a microfabricated array of soft polydimethylsiloxane (PDMS) micropillars in a chip (9 x 9 mm) and, from the displacement of the pillars, the force exerted by the worm is calculated using a modified form of the Timoshenko beam deflection theory. At any given time a single worm is in contact with 6-10 pillars, primarily depending on pillar spacing. Interestingly, the force varies depending on which position along the worm length is measured, with the maximum force exerted from the middle part of the worm body. This result agrees with theoretical analysis that predicted that maximum force is generated near the middle part of the body (Shen et al., 2012). The maximum force measured was 31.3 μN (this was obtained with pillars arranged in a hexagonal lattice; a lower maximum force of 18.9 μN was obtained with pillars arranged in a square lattice; in each case the pillar to pillar distance was 140 μm). The recordings could also be easily analyzed to derive speed, amplitude and wavelength. So far, there is only one report in which this method has been used to measure forces exerted by muscle mutants: Etheridge et al. (2015) reported decreased peak forces from unc-52ts and unc-112ts adults at the restrictive temperature.
Although the forces measured by the AFM (Atomic Force Microscopy) and micropillar methods are close (31-50 μN), they do not agree with measurements of forces exerted by single worms crawling on agar surfaces. For example, Rabets et al. (2014), after measuring drag forces exerted on worms as they move along an agar surface, used “resistive force theory” to calculate the force generated by a single crawling worm as ≈5 μN, an order of magnitude lower than the methods noted above. Using kinematic data and a hydrodynamic model based on lubrication theory, Shen et al. (2012) calculated a much lower bending force of 85-89 nN. The reasons for this wide variation in measured forces is not clear but may certainly depend on variation in methodology. The AFM method measures an “escape force”, the micropillar method measures deflection of pillars, but the other studies involved measurement or calculation of force on a wet viscoelastic agar gel. Moreover, various studies, using different methods, have reported that the bending force of individual worms in liquid is ≈1-3 nN (Krajacic et al., 2012; Shen et al., 2012; Kuo et al., 2014). It is clear that nematodes generate much higher bending forces to crawl on a surface than to swim in liquid. Despite the method used, most of these methods have been used to distinguish wild type from mutants. It is hoped that more of this analysis will be reported for phenotyping muscle mutants in the future.
Multiple computer programs (Nemo, Worm Tracker 2.0, Parallel worm tracker, Multi Worm Tracker, The Tracker, etc.) for tracking and measuring crawling of C. elegans on a solid surface have been described (WormBook chapter Keeping track of worm trackers). There is also a program (CeleST) available for analyzing swimming (Restif et al., 2014). These programs quantify different aspects of locomotion such as posture, directional changes, curvature of the sinusoidal shape, and velocity. The programs have been used to analyze the locomotion of wild type and various mutants (e.g., Brown et al., 2013; Swierczek et al., 2011; Koren et al., 2015). However, the main focus of these studies has been on neuronal mutants, only a few muscle uncs have been analyzed. This type of analysis clearly has potential for understanding muscle gene function.
Due to the small size of C. elegans and consequent inability to isolate muscle cells or groups of muscle cells, it has not been possible to conduct measurements of muscle kinetics as is conducted on individual muscle fibers from vertebrate muscle. However, recently some kinetic parameters have been measured by optogenetically controlling muscle contraction of whole nematodes. In a report by Hwang et al. (2016) (Figure 7D), a worm strain was used in which channelrhodopsin-2 is expressed in cholinergic motor neurons. Illumination was used to induce contractions in body wall muscles. A measure of muscle contraction state is total body area normalized to the length of the animals. Using image processing, the change in body area vs. time when light is on and off was plotted.
Results were obtained for wild type and mutants in genes encoding 15 sarcomere proteins. Curve fitting with one-phase decay and one-phase association models were used to calculate rate constants for contraction and relaxation, respectively. Many of the mutants were defective in these and related parameters. unc-54(s74) was the only mutant tested that showed a decrease in the rate of contraction. This is perhaps expected since s74 is a missense mutation in the ATP binding site that is likely to result in reduced ATPase and motor velocity and a slower contraction/relaxation cycle (Moerman et al., 1982; Moerman and Fire, 1997). Increased relaxation rates observed in two of the mutants, unc-27 and unc-22, might be explained from what is known or speculated about their roles in muscle activity. UNC-27 is one of four troponin I isoforms (Burkeen et al., 2004) and in vertebrate striated muscle troponin I is known to inhibit the interaction of myosin heads with thin filaments (Perry, 1999). unc-22 encodes twitchin (see below), and both the loss-of-function twitching phenotype, and the presence of a protein kinase domain similar to myosin light chain kinase suggests a role for twitchin in regulating muscle contraction. Prior work in Aplysia and Mytillus suggested that twitchin inhibits the rate of relaxation (Probst et al., 1994; Siegman et al., 1998; Funabara et al., 2007). It is noteworthy that the rate constants for relaxation are increased by two unc-22 alleles, e66 and e105: e66 leads to disorganized sarcomeres, whereas e105 has normal organization of sarcomeres (Matsunaga et al., 2015). This suggests that twitchin has two separable functions, one structural and one regulatory.
There is growing recognition that in mammalian striated muscle, a number of Z-disk and M-line proteins, including LIM domain proteins (e.g., MLP, FHL2), translocate to the nucleus in response to mechanical stimuli or extracellular signals, and once inside the nucleus, influence gene transcription (Lange et al., 2005; Gautel, 2008). In C. elegans, some M-line and dense body proteins are also found in the nucleus (ALP-1, FRG-1, UNC-95, UNC-97, UNC-98, ZYX-1). However, the functional significance of this nuclear localization is unknown. The most significant finding would be to show that proteins from the cell surface shuttle to the nucleus. As noted below, FRAP experiments indicate when either ZYX-1::GFP (Lecroisey et al., 2013) or UNC-98::GFP (R.K. Miller and G.M. Benian, unpublished data) are bleached in the nucleus, the nuclear signal re-appears, presumably from protein originating from the cytoplasm. However, whether this protein originates from near the cell surface (dense bodies and M-lines) or from cytoplasmic pools is unknown. A definitive experiment would be to use photoactivatable GFP tagged proteins, activate them at the cell surface and then monitor their possible appearance in the nucleus. Such an experiment has not been reported.
In transgenic worms, translational GFP fusions of full-length UNC-97 (Hobert et al., 1999), UNC-98 (Mercer et al., 2003), UNC-95 (Broday et al., 2004), and ZYX-1 (zyxin) (Lecroisey et al., 2008; Lecroisey et al., 2013) show localization to M-lines, dense bodies, and nuclei. However, antibodies developed to UNC-98 (Mercer et al., 2003), to UNC-97 (Miller et al., 2006), and to UNC-95 (Qadota et al., 2007), when used in immunofluorescent experiments, failed to localize to nuclei under normal conditions. Nevertheless, anti-UNC-98 reacted to nuclei when a non-standard fixation method was used, or when UNC-98 was overexpressed (Mercer et al., 2003). Additional support that endogenous UNC-98 and UNC-97 reside in nuclei was obtained during purification of native thick filaments reported in Miller et al., (2006): nuclear-enriched fractions contained UNC-98 and UNC-97 detectable by Western blot. In the Y2H system, when either UNC-98 (Mercer et al., 2003) or UNC-97 (Mackinnon et al., 2002) are fused to the GAL4 DNA binding domain, they can activate transcription, suggesting that UNC-98 and UNC-97 may activate transcription in vivo. By testing deletion derivatives of UNC-98::GFP, the N-terminal 110 residues of UNC-98 are sufficient for nuclear localization (Mercer et al., 2003). A similar approach by Norman et al., (2007) indicates that the LIM2 and LIM3 domains are required for nuclear localization of UNC-97 (PINCH).
Nuclear localization has also been found for the dense body proteins ALP-1 (McKeown et al., 2006) and FRG-1 (Liu et al., 2010). alp-1 encodes four isoforms, one is ALP-like (ALP-1A), and three are Enigma-like (ALP-1B, -1C, -1D) (McKeown et al., 2006). Use of GFP translational fusions demonstrates a complex pattern of expression of genes encoding these proteins in embryos and adults, and localization to dense bodies and nuclei of muscle and hypodermal (epithelial) cells. In fact, ALP-1 is one of the few integrin adhesion site proteins showing strong localization to embryonic muscle (ALP-1A) and hypodermal cell (ALP-1B, C, D) nuclei. FRG-1 is the C. elegans ortholog of the human facioscapulohumeral muscular dystrophy region gene 1 (FRG1) (Liu et al., 2010). Antibodies to FRG-1 localize both to dense bodies, and to muscle cell nuclei, concentrated in the nucleoli. FRG-1 bundles F-actin in vitro. Overproduction of FRG-1 from the myo-3 muscle specific promoter resulted in no obvious defects in muscle structure or nematode locomotion. However, when overexpressed from its own promoter, Liu et al. (2010) observed disruption of some muscle-muscle lateral junctions and/or absence of some muscle cells in the ventral but not dorsal musculature.
In contrast to most other Pat genes that encode components of the integrin adhesion sites, PAT-9 is exclusively localized to nuclei (Liu et al., 2012). This was determined by both antibodies to PAT-9 and use of a PAT-9::GFP translational fusion in transgenic animals. In addition to localizing to embryonic, larval, and adult body wall muscle cell nuclei, PAT-9 also localizes to germline nuclei of the syncytial gonad. Localization is confined to the DAPI poor, presumably nucleolar region. PAT-9 is a 470-residue long protein containing three C2H2 Zn fingers and one predicted nuclear localization sequence (NLS). This NLS was confirmed by transgenic experiments. Given that pat-9, like deb-1, is a class III Pat gene required specifically for the formation of actin filaments (Williams and Waterston, 1994), one hypothesis is that PAT-9 is a nuclear C2H2 zinc finger transcription factor that regulates the expression of genes critical for assembly of dense bodies or thin filaments. However, a yeast one-hybrid screen (Liu et al., 2012) showed that PAT-9 is bound to the promoters of five genes (daf-3, tbx-2, cog-1, let-7, and mir-76). Three of these five genes (let-7, daf-3, and tbx-2) are expressed in body wall muscle by promoter (WormBase) and/or SAGE analysis (Meissner et al., 2009). ChIP assays performed by Liu et al. (2012) confirmed that PAT-9 binds to the promoters of tbx-2 and daf-3. An additional gene tested by ChIP was frg-1, which, as noted above, encodes a protein localized to dense bodies and muscle cell nuclei. PAT-9 was also found bound to the promoter of frg-1.
Two additional M-line and dense body proteins have recently been reported. These are PKN-1 (PKN) and PXL-1 (paxillin). PKN-1 is the C. elegans ortholog of protein kinase N (PKN), an effector of RhoA. The pkn-1 promoter is expressed in body wall muscle, and a GFP::PKN-1 fusion protein localizes to M-lines and dense bodies (Qadota et al., 2011). An intragenic deletion of pkn-1 or heat shock-induced overexpression of the protein kinase domain has normal sarcomere structure, but displays an unusual “loopy Unc” phenotype, which has been reported in many mutants of neuronal genes. The results of mosaic analysis and body wall muscle over-expression of the kinase domain (using the myo-3 promoter) suggest that this loopy phenotype is due to expression of PKN-1 in body wall muscle.
Warner et al. (2011) report that PXL-1 is the nematode ortholog of paxillin, a well-known component of integrin adhesion sites of vertebrates. The protein is expressed in both body wall and pharyngeal muscles. Based on antibody staining and a GFP fusion protein, PXL-1 localizes to dense bodies and M-lines in body wall muscle, and to ring-shaped structures near the sarcolemma in pharyngeal muscle corresponding to podosome-like sites of actin attachment. A pxl-1 intragenic deletion mutant shows L1 arrest with paralyzed pharyngeal muscle and ultimately lethality, probably due to an inability to feed. The lethality can be rescued by expressing PXL-1 only in the pharynx. Rescued animals develop into adults that show normal locomotion and normal body wall muscle sarcomere organization. Therefore, PXL-1 is not required for the structure or function of body wall muscle. PXL-1 was found to interact with numerous proteins, including UNC-95. In body wall muscle of an unc-95 null mutant, PXL-1 is mis-localized. In body wall muscle, pxl-1 is likely to be redundant with a second LIM domain containing protein, LIM-8, since in the pharyngeally-rescued pxl-1 deletion mutant, RNAi for lim-8 results in slow moving animals with mildly disorganized sarcomeres. A lim-8 null mutant has normally organized sarcomeres (Qadota et al., 2007).
Meissner et al. (2009) reported that an RNAi screen of ≈3300 muscle-expressed genes identified 108 new genes that are important for sarcomere assembly and/or maintenance. Actually, two screens were performed, one for new Pat genes, and one for new genes that when knocked down result in abnormal sarcomere organization of adult muscle. In the Pat screen, four new Pat genes were discovered: T27B1.2, F31D5.3, T28B4.3, and F25B3.6. F31D5.3 was independently identified as a Pat gene and re-named cpna-1 (Warner et al., 2013; see below). T27B1.2 was later identified as pat-9 (Liu et al., 2012; see above). The second RNAi screen involved looking for defects in myosin localization in adults by using a GFP::MHC A expressing strain. Myosin mis-localization was observed for RNAi knockdown of 104 genes not previously known to be involved in myofibrillar organization. Many of these genes have human homologs for which little or nothing is known.
In a related study, Meissner et al. (2011), using transgenic animals expressing GFP-tagged proteins, determined the sub-cellular localization of 227 muscle-expressed proteins. For most of these, no previous information on sub-cellular localization was known. The authors described 14 different sub-cellular localization patterns within body wall muscle cells (e.g., thick and thin filaments plus or minus dense bodies; dense bodies, M-lines, and attachment sites; dense bodies and attachment sites; dense bodies, cytoplasm (plus or minus M-lines), nucleus; sarcoplasmic reticulum; nucleolus; nucleus; etc.). Localization patterns were obtained for 37 of the 108 new genes identified from their previous RNAi study (Meissner et al., 2009). The authors rightly point out that it is somewhat uncertain whether the location of a given protein determined by their study actually reflects the location of a given endogenous protein in vivo. This is because the proteins had the ≈200 residue GFP molecule attached to their C-termini that could possibly interfere with normal localization and function. In addition, the transgenic approach used is an overexpression situation that could potentially result in mis-localization. Also, all of the proteins were expressed from a single muscle-specific promoter (from T05G5.1), not the genes’ own promoter. It will be important to verify these locations by antibodies to endogenous proteins and/or the use of CRISPR to tag the endogenously expressed gene products.
Most recently, Etheridge et al. (2015) have reported results of a search for muscle phenotypes by RNAi of a large number of C. elegans homologs of proteins that comprise the human integrin-adhesome. The authors knocked down, by RNAi in adults, 113 C. elegans genes that are homologs of the 151 human integrin-adhesome proteins and assessed the structure of sarcomeres by the localization of GFP::MHC A, and the structure of integrin attachment sites by localization of UNC-95::GFP. Fifty-five (49%) showed defects in thick filaments, and 22 (19.5%) showed defects in attachment structures. Nine of these genes had not previously been found to have one or more of these defects. What is more impressive from this study, however, is that RNAi of 102 of the 113 targets (90%) resulted in defects in mitochondrial structure as assessed by imaging GFP tagged mitochondria for fragmented or disorganized mitochondrial networks. Moreover, the authors found that the maximum rates of mitochondrial ATP production from purified mitochondria were decreased in either unc-52ts or unc-112ts mutants. The analysis of mitochondrial defects is an important contribution to the nematode muscle field. The effects of reduced expression or mutation of sarcomeric and attachment proteins on the organization and function of mitochondria is an underappreciated and understudied area. The authors also knocked down 47 genes that encode new attachment site proteins, as described above by Meissner et al. (2011). Similarly, 46 of the 47 genes (98%) resulted in defects in mitochondria, and 15 of the 47 (32%) resulted in defects in thick filament organization and/or the attachment structures. Apparently, reduced expression of mitochondrial proteins can result in disorganization of the myofilament lattice: of the 25 proteins determined to be localized to mitochondria by Meissner et al. (2011), four (mdh-2, R119.3, T09A5.5, T10B11.6) result in disorganization of thick filaments when knocked down by RNAi (Meissner et al., 2009).
Using a new method to enrich for antigens of low abundance from an embryo extract, Takeda et al. (2008) have isolated 35 monoclonal antibodies that recognize P granules, muscle, the pharynx, and hypodermal cells. Six of these “KT” series antibodies recognize structures in body wall muscle or the basement membrane. Several have novel localization patterns, including KT12 that seems to recognize the polar regions of A-bands, even more extreme than MHC B. It should prove useful to identify the corresponding antigens.
Thus, through the studies of Meissner et al. (2009; 2011), Etheridge et al. (2015), and Takeda et al. (2008), we have many new muscle genes/proteins to study. It will be informative to now characterize the phenotypes of loss-of-function mutants, especially null mutants, for these genes that have been so far only identified through RNAi. It will also be important to study their genetic and biochemical interactions with each other and already known components of muscle. For example, protein-protein interactions could be studied by Y2H assays and binding assays with purified components. In addition, using antibodies or tagged proteins, protein complexes could be identified, and the stoichiometries of the proteins involved can be determined by SILAC-based immunoprecipitations and mass spectrometry.
Biochemical studies indicate that myosin heads do not fold spontaneously and require additional proteins (Chow et al., 2002; Resnicow et al., 2010). The conserved protein UNC-45 was the first myosin head chaperone discovered, and it was first revealed from studies of C. elegans. The gene was identified by a temperature sensitive allele, e286, which displays sarcomere disorganization and reduced numbers of thick filaments (Epstein and Thomson, 1974). When grown at the restrictive temperature, e286 animals show decreased accumulation of MHC B, but not paramyosin or MHC A (Barral et al., 1998). Interestingly, the normal differential localization of MHC A to the middle, and MHC B to the polar ends of the thick filaments is lost: in e286, both MHC A and B are found throughout the thick filaments.
The UNC-45 protein is 961 residues long and consists of 3 regions: a TPR (tetratricopeptide repeat) region (3 TPR (34 residue long) repeats), a central unique region, and a UCS (UNC-45, CRO1, She4p) domain (Barral et al., 1998; Venolia et al., 1999). The UCS domain was defined by sequence homology to two fungal proteins that functionally interact with myosin. The unc-45 temperature sensitive mutations are amino acid substitutions in the UCS domain whereas Pat embryonic lethal mutants carry stop codons upstream of the UCS domain (Venolia and Waterston, 1990; Barral et al. 1998). The domains of UNC-45 are functionally distinct in vitro and in vivo. The TPR region binds to heat shock protein 90 (Hsp90), and the central region and UCS domain bind to myosin heads (Barral et al., 2002). Moreover, UNC-45 can inhibit the thermal aggregation of myosin heads in vitro, suggesting that UNC-45 chaperones the myosin head (Barral et al., 2002). In adult body wall muscle antibodies to UNC-45 localize the protein to the polar regions of the A-band, co-localizing with MHC B (Ao and Pilgrim, 2000; Gazda et al., 2013). It is curious as to why UNC-45 does not colocalize with MHC A, whose myosin heads presumably also require a chaperone.
Clever single molecule experiments using AFM provide nearly direct evidence that UNC-45 promotes the folding of the myosin head (Kaiser et al., 2012). A polyprotein consisting of eight tandem copies of an Ig domain from human titin (with known AFM force-displacement profiles) attached to either full length myosin or myosin S1 heads (from rabbit skeletal muscle) was mechanically stretched three consecutive times, separated by relaxation for 10 seconds to allow refolding of the Ig domains. Normally, a set of Ig domains will spontaneously refold, as indicated by observing the Ig domain sawtooth pattern on a force vs. displacement graph for each consecutive stretch. When performed with the myosin-Ig set polyprotein, the sawtooth pattern was absent in the second and third pulls, suggesting that the unfolded myosin head interferes with refolding of the Ig domains. However, when repeated with inclusion of UNC-45 (mouse UNC-45b), the sawtooth pattern was seen in four consecutive stretches, indicating that the Ig domains serve as a “sensor” for myosin head refolding promoted by UNC-45 (Kaiser et al., 2012).
The myosin-binding UCS domain alone is sufficient to rescue both lethal unc-45 null (Pat) mutants, and ts loss-of-function unc-45 mutants. Removal of the Hsp90-binding TPR domain from portions of UNC-45 that contain the UCS does not affect this rescue (Ni et al. 2011). Titration experiments demonstrate that, on a per mole basis, UCS has greater activity in vivo than full-length UNC-45, suggesting that full length UNC-45 is inhibited by either the TPR domain or its interaction with Hsp90. Using purified recombinant proteins it can be shown that Hsp90 and UNC-45 compete for interaction with myosin. Therefore, Hsp90 has an inhibitory role in relation to the activity of UNC-45 in promoting myosin head folding and/or thick filament assembly (Ni et al., 2011).
The sole cytosolic Hsp90 in C. elegans is DAF-21 and it is 74% and 76% identical to human Hsp90α and β, respectively (Birnby et al., 2000). RNAi knockdown of daf-21 in a strain expressing GFP::MYO-3 results in abnormally organized A-bands and aggregates of MYO-3 (MHC A) (Gaiser et al., 2011). Moreover, both daf-21 RNAi animals and the loss-of-function mutant daf-21(p673) show decreased motility in a thrashing assay (Gaiser et al., 2011). Using a traditional transgenic approach in which YFP::DAF-21 is overexpressed, YFP::DAF-21 localized primarily to the I-band. Interestingly, FRAP indicates that because fluorescence recovered very rapidly and completely (t1/2 ≈1 sec), the authors conclude that DAF-21 is freely diffusible and only transiently associated with the I-band.
UNC-45 homologs are found in all metazoa (Price et al., 2002); Drosophila and C. elegans have one UNC-45 gene expressed in all cells; vertebrates have two UNC-45 genes, one expressed in striated muscle (UNC-45B) and one expressed in all cells (UNC-45A). C. elegans UNC-45 not only chaperones body wall muscle myosins, it also chaperones NMY-2, a non-muscle myosin II (Kachur et al., 2004; Kachur et al., 2008). Maternally contributed UNC-45 acts with NMY-2 during embryonic polarity establishment, cytokinesis, and germline cellularization (Kachur et al., 2008). In early embryos UNC-45 and NMY-2 co-localize at cell boundaries (Kachur et al., 2004). A Y2H library screen with UNC-45 revealed interaction with NMY-2 and with HUM-2, a class V myosin (Kachur et al., 2004). Therefore, UNC-45 encoded by its single gene, interacts with, and likely chaperones, both conventional (UNC-54, NMY-2) and unconventional (HUM-2) myosins.
Although we do not know how UNC-45 aids the folding of the myosin head, we do have some insight into the complex mechanism by which UNC-45 aids thick filament assembly. It is very likely that UNC-45 regulates the levels of myosins in coordination with degradation of myosins via the ubiquitin proteasome system (Landsverk et al., 2007). Both loss-of-function mutants and transgenic overexpression of unc-45 lead to decreased myosin accumulation, decreased thick filament assembly, and decreased nematode locomotion (Landsverk et al., 2007). By using different segments of UNC-45, it was determined that the overexpression phenotype depends on whether UNC-45 contains the UCS domain (Ni et al., 2011). A mechanism compatible with these results is that the concentration of soluble complexes of UNC-45 and mis-folded myosin is rate-limiting for accumulation of normally folded myosin and its assembly; too low or too high concentrations of these complexes lead to shifting to unfolded myosin and its ubiquitinylation and proteasomal degradation (Landsverk et al., 2007; Epstein and Benian, 2012).
The first crystal structure of an UNC-45 type molecule was obtained for Drosophila UNC-45 (Lee et al., 2011). Although present in the crystal, the TPR domain could not be “seen”, exhibiting either disorder or flexibility relative to the central and UCS domains. The overall shape of the protein is a “horseshoe” with the central region making up the short leg. Interestingly, the entire central and UCS domains consist nearly entirely of 17 armadillo (ARM) repeats, each consisting of 2-3 α-helices. The crystal structure of C. elegans UNC-45 (recombinant protein expressed in E. coli) shows that UNC-45 forms a linear multimer (Gazda et al., 2013). The length of the repeating unit in this multimer (17.0 nm) is similar to the repeating unit in the staggered arrangement of myosin heads in a thick filament (closest distance between adjacent double heads is 14.3 nm). Although staggering of myosin heads on the surface of thick filaments is dictated by the stagger of 98 residues of parallel myosin rods to optimize charge-charge interactions (McLachlan and Karn, 1982), UNC-45 multimers may help stabilize this arrangement during thick filament and sarcomere assembly (Figure 8A). UNC-45 multimers may also be recruited to and assemble onto established thick filaments to assist the refolding of damaged myosin heads.
|
Figure 8. Model for the action of UNC-45 in sarcomere assembly and sarcomere repair. (a) UNC-45 (orange) forms a multimer with periodicity matching the display of myosin heads on the thick filament surface. As thick filaments are forming, or damaged myosin heads are being repaired in mature muscle, the UNC-45 multimer binds to the thick filament allowing hydrolysis of ATP but locking myosin into an actin-bound conformation, without powerstrokes taking place. (b) Addition of Hsp90 results in release of UNC-45 from the thick filament in which myosin heads are properly folded, and powerstokes are free to occur. (c) The absence of UNC-45 results in the disorganization of the sarcomere, which is then targeted for proteasomal degradation. (Reprinted, with permission, from Nicholls et al. 2014)
If UNC-45 promotes the folding of myosin heads and/or maintains the folded state, one would predict that the presence of UNC-45 would enhance the activity of the myosin head. However, surprisingly, Nicholls et al. (2014) found the opposite effect. Addition of bacterially expressed mouse UNC-45B to rabbit skeletal muscle myosin or myosin subfragment-1 (S1) inhibited the ability of myosin to move fluorescently labeled actin filaments in a classical actin filament gliding assay. However, UNC-45 did not inhibit myosin ATPase activity, nor did UNC-45B bind to F-actin. Although the TPR domain of UNC-45 can recruit Hsp90, the authors found that Hsp90 on its own did not affect actin gliding, but when Hsp90 was added after the UNC-45-induced halt, Hsp90 reversed it. This Hsp90 reversal of UNC-45 induced halt of myosin motor activity depended on the ATPase activity of Hsp90 as it was inhibited by geldanamycin. Therefore, the authors conclude that UNC-45 inhibits the power stroke but not the hydrolysis of ATP. They go on to further speculate that UNC-45’s inhibition of myosin head activity has crucial roles both during formation of sarcomeres and during repair of sarcomeres. That is, while UNC-45 assists the myosin head to attain its native conformation, it prevents powerstrokes that would otherwise be damaging to the assembly. Once the myosin head is properly folded, the inhibited state of the myosin head is relieved when Hsp90 binds to UNC-45 and releases UNC-45 from the myosin head (Figure 8). This model needs to be confirmed by showing that nematode UNC-45 has a similar activity.
Mutations in the unc-23 gene result in detachment of body wall muscle cells from the hypodermis beginning during mid-larval development (Waterston et al., 1980; Plenefisch et al., 2000). This detachment only occurs in the front or “head” region of the animal, and results in adults with a thinner head region that is bent. Interestingly, this bent-head phenotype only occurs when unc-23 mutants are grown on plates; when grown in liquid culture, there is no detachment or bent heads. Nevertheless, when liquid-grown unc-23 mutants are subjected to mechanical stress by gently rolling them under a coverslip, the muscle cells do detach (Rahmani et al., 2015). As worms need to overcome higher surface tension when moving on agar than in liquid, this suggests that the function of UNC-23 is to resist mechanical force in the attachment of muscle cells to hypodermis.
unc-23 encodes potentially 3 isoforms up to 458 residues long (Rahmani et al., 2015). At its C-terminus is a 45 residue BAG domain, which UNC-23 shares with human BAG-2, a member of the Bcl-2 associated athanogene (BAG) family of molecular chaperone regulators. In other members of this family, the BAG domain binds to and modulates the activity of the ATPase domain of the heat shock cognate protein 70, Hsc70, thus inhibiting its chaperone activity. A rescuing UNC-23::GFP fusion protein is expressed throughout development and is expressed in both body wall muscle and hypodermis (Rahmani et al., 2015). In body wall muscle UNC-23::GFP is localized to dense bodies and M-lines near the sarcolemma, and in hypodermal cells in a pattern similar to intermediate filaments and also in nuclei and nucleoli.
An unc-23 suppressor screen resulted in isolation of 7 extragenic suppressor strains, two of which are alleles of hsp-1, which encodes the nematode ortholog of Hsc70. Both alleles of hsp-1 are amino acid substitutions in the ATPase domain of the HSP-1 protein. Using the BAG domain of UNC-23 as bait, a screen of a Y2H library yielded multiple clones representing HSP-1, and the minimal region of interaction contained part of the ATPase domain of HSP-1. In support of the genetic and Y2H evidence that UNC-23 and HSP-1 interact, Papsdorf et al. (2014) have shown that UNC-23 and HSP-1 interact in vitro, and have also demonstrated that UNC-23 acts as a nucleotide exchange factor. Moreover, Papsdorf et al. (2014) show that overexpression of HSP-1 (in transgenic animals expressing hsp-1p::CFP::HSP-1) results in a phenotype similar to the bent-head phenotype of unc-23 loss-of-function mutants. This suggests that the level of the HSP-1 chaperone needs to be tightly regulated to maintain the proper folding of substrates in muscle or hypodermis to maintain the strength of attachment structures. It is known that Hsc70 is also regulated by Hsp40. Interestingly, RNAi knockdown of one of the Hsp40 proteins in C. elegans, DNJ-13, suppressed the unc-23 muscle phenotype (Papsdorf et al., 2014). Although UNC-23 is expressed in both muscle and hypodermis, the critical tissue is likely to be the hypodermis; expression of UNC-23 only in muscle was not sufficient to rescue the bent-head phenotype of unc-23 mutants (Rahmani et al., 2015).
Thus, these seminal studies by Rahmani et al. (2015) and Papsdorf et al. (2014) demonstrate a role for a molecular chaperone (HSP-1) and its regulators, UNC-23(BAG-2) and DNJ-13 (Hsp40), in the maintenance of muscle adhesion complexes so that they function optimally in the transmission of mechanical force generated in muscle, through the hypodermis, and ultimately to the cuticle. It will be interesting to determine which proteins of hypodermal cells, and dense bodies and M-lines in muscle cells, are the substrates of this folding complex—perhaps the folding of only one or a few proteins is critically involved.
Finally, it should be mentioned that HSP-25 was the first molecular chaperone reported to be associated with the sarcomere in C. elegans (Ding and Candido, 2000). HSP-25 is localized to dense bodies and M-lines (Ding and Candido, 2000). HSP-25 is one of 16 “small heat shock proteins (smHSPs)” in the C. elegans proteome (Ding and Candido, 2000). smHSPs are found in all kingdoms of life and although they are incapable of catalyzing the refolding of protein substrates, they are able to bind partially unfolded proteins and hold them in a folding-competent state for interaction with other chaperones which can catalyze refolding. The localization of HSP-25 to integrin adhesion sites suggests that HSP-25 may be involved in the maintenance, turnover, or assembly of these structures. Alternatively, the M-lines and dense bodies might represent storage sites from which HSP-25 can exit to assist folding of damaged proteins elsewhere in muscle. Ding and Candido (2000) showed that an affinity column of HSP-25 is capable of pulling out DEB-1 and ATN-1 from worm extracts. However, whether these proteins interact directly was not demonstrated. Further, the authors reported that hsp-25 RNAi resulted in no obvious phenotype (Ding and Candido, 2000). Although an analysis of hsp-25 mutants has not been reported, there is a nonsense mutation in this gene available in the Million Mutation Project (MMP) collection (Thompson et al., 2013).
The M-line proteins UNC-96 and UNC-98 interact with CSN-5 (Miller et al., 2009). Interactions were identified by a Y2H library screen and confirmed by biochemical methods. CSN-5 is a component of the highly conserved “COP9 signalosome complex” that is found in multiple organisms to regulate protein stability, usually through SCF ubiquitin ligases (Cope and Deshaies, 2003; Schwechheimer, 2004). Anti-CSN-5 antibody co-localizes with paramyosin at A-bands in wild type, and co-localizes with abnormal accumulations of paramyosin found in unc-98, unc-96, and unc-15 mutants. RNAi knock down of csn-5 results in an increase in the level of UNC-98 protein and a slight decrease in the level of UNC-96 protein, suggesting that CSN-5 promotes the degradation of UNC-98 and that CSN-5 stabilizes UNC-96. In unc-15 and unc-96 mutants, CSN-5 protein is reduced, implying the existence of feedback regulation from myofibril proteins to CSN-5 protein levels. This implicates for the first time CSN-5 or the COP9 signalosome in myofibrillar organization. These findings are consistent with the ubiquitin proteasome system being required for muscle protein turnover in vertebrate muscle, mediated by the muscle specific ubiquitin ligase Atrogin-1 and the MuRF family (Muscle specific RING Finger proteins) (Bodine et al., 2001; Gomes et al., 2001).
Moreover, in C. elegans muscle, the RING finger protein RNF-5 is localized to dense bodies and regulates the levels of UNC-95 (Broday et al., 2004). RNF-5 and UNC-95 interact by Y2H (Didier et al., 2003): heat shock induced overexpression of RNF-5 results in a reduction in UNC-95::GFP, and this reduction depends on the presence of an active RING finger domain in RNF-5; in contrast, RNAi mediated knockdown of rnf-5 results in an increase in UNC-95::GFP (Broday et al., 2004).
Both UNC-89 and its vertebrate homolog, obscurin, play important roles in regulating protein turnover in muscle, again, through the ubiquitin/proteasome system. Two segments of UNC-89 (Ig2-Ig3 and 1/3 interkinase-Ig53-Fn2) interact with MEL-26, a substrate recognition protein for cullin 3 (Wilson et al., 2012). Cullins, first discovered in C. elegans (Bosu and Kipreos, 2008), are conserved scaffolds for assembly of the ubiquitin protein degradation machinery, including E3 ubiquitin ligases. By antibody staining, MEL-26 and UNC-89 partially colocalize at sarcomeric M-lines. This was the first time that a component of a cullin complex had been reported in the sarcomere. Loss-of-function (lof) or gain-of-function (gof) mutations of mel-26 result in disorganization of myosin thick filaments similar to that found in unc-89 mutants. It had been reported previously that in early C. elegans embryos, a target of the CUL-3/MEL-26 complex is the microtubule-severing enzyme katanin (MEI-1) (Furukawa et al., 2003; Pintard et al., 2003; Xu et al., 2003). Lof or gof of mei-1 also result in disorganization of thick filaments similar to unc-89 mutants. Genetic data indicated that at least some of the mel-26 lof phenotype in muscle can be attributed to increased microtubule severing activity of MEI-1. The level of the MEI-1 protein is reduced in an unc-89 mutant, and MEI-1 is normally degraded by the proteasome. A model proposes that the interaction of UNC-89 with MEL-26 inhibits the activity of the CUL-3/MEL-26 complex from promoting the ubiquitin-mediated degradation of MEI-1. MEI-1 severs microtubules, and this activity is in some way required for thick filament organization in muscle (Wilson et al., 2012).
Similarly, Lange and colleagues (2012) have reported in their studies on the obscurin knockout mouse that degradation of small ankyrin 1.5 (sAnk1.5) is dependent upon obscurin, and is promoted by a cullin 3 substrate recognition protein, KCTD6. Thus, independent studies point to evolutionarily conserved mechanisms by which UNC-89 or obscurin regulate ubiquitin mediated protein degradation in muscle.
Etheridge et al. (2012) have reported a fascinating role for integrin adhesion sites in blocking activation of the proteases called calpains. Moreover, calpains were shown to be required for the maintenance of integrin adhesion sites. RNAi in adults for any one of 14 adhesion site proteins (PAT-2, PAT-3, PAT-4, PAT-6, UNC-52, UNC-97, UNC-112, TLN-1, ZYX-1, UNC-82, ATN-1, DEB-1, CDC-42, and UIG-1) results in decreased levels of cytosolic LacZ, a reporter of muscle protein degradation. Interestingly, RNAi knockdown of one protein, UNC-89, did not result in loss of cytosolic LacZ (UNC-89 may have a role in inhibiting the ubiquitin proteasome system at the M-line (see above)). Similar induction of cytosolic LacZ degradation was observed when ts mutants of either unc-52 or unc-112 adults were grown at the restrictive temperature. Moreover, the effect was not specific for LacZ; degradation of cytosolic GFP or cytosolic DsRed was also observed in unc-97(RNAi) and unc-112(RNAi). Using several chemical inhibitors of the ubiquitin proteasome system or autophagy, and mutants in autophagic pathways or cell death (caspases), the authors found that degradation of cytosolic LacZ could not be blocked. However, they were able to block degradation induced by either unc-52ts or unc-112ts, by treatment with chemical inhibitors of calpains, or RNAi for any one of five calpain encoding genes (clp-1, clp-4, tra-3, clp-6, and clp-7). Therefore, they conclude that calpains are the proteases that are activated when integrin attachment complexes are disrupted. Although this study is thorough and rather compelling, one criticism is that it heavily relies on the monitoring of degradation of an overexpressed artificial cytosolic protein. However, the authors did show by quantitative Western blot that an endogenous dense body protein, DEB-1 (vinculin), is also degraded in unc-112ts and this degradation can be blocked by a calpain inhibitor. Finally, RNAi against any of the calpain genes resulted in defects in adult muscle, either when RNAi is performed in adults or when RNAi is performed for several generations, including disruption in myofilament lattice array and, at least for clp-1, disruption of attachment complexes. Their conclusion is that calpains are required to maintain integrin attachment complexes. This is consistent with the role of calpains in maintaining focal adhesions in vertebrate tissue culture cells (Lebart and Benyamin, 2006). A possible mechanism might be that the calpains contribute to the removal of damaged proteins at the attachment complexes.
Loss of proteostasis has emerged as a hallmark of the cellular aging process (Taylor and Dillin, 2011). The failure of proteins to fold or to maintain folded states can result in the formation of toxic aggregates and loss of essential cellular functions. Muscle aging, also referred to as sarcopenia, is characterized by the accumulation of misfolded proteins contributing to the loss of skeletal muscle function and mass. In C. elegans, progressive locomotory impairment has been described during aging and proposed to be the consequence of a decline in muscle function (Herndon et al., 2002). The authors demonstrate that aged muscle shows a progressive decrease in sarcomere organization. In addition, using aggregation-prone PolyQ peptides expressed in muscle cells, Morley et al. (2002) demonstrated an age- related increase of protein aggregation and proteotoxicity that correlates with motility decline. These results suggest that maintainance of sarcomere structure and proteostasis, both processes involving molecular chaperones and proteases, are essential for the preservation of correct muscle function. Moreover, because aging as well as the control of proteostasis are cell nonautonomous processes (reviewed in van Oosten-Hawle and Morimoto, 2014), molecular chaperones and muscle proteases constitute potential targets to prevent age related-loss of muscle function and potentially an increase in overall healthspan.
Muscle sarcomeres contain a number of giant polypeptides (>700 kDa), consisting of multiple copies of immunoglobulin (Ig) and fibronectin type 3 (Fn) domains, and one or even two protein kinase domains near their C-termini. These are all related to the largest polypeptide known, titin, which is found in vertebrate striated muscles. Much is currently known about the function of vertebrate titin, including its role in myofibrillar assembly, muscle elasticity, and muscle specific signaling pathways. Mutations in the human titin gene are responsible for a number of myopathies and cardiomyopathies. C. elegans muscle contains three such giant protein kinases: twitchin (754,000 Da) located at the outer regions of the A-band, TTN-1 (2.2 MDa) located in the I-band, and UNC-89 (multiple isoforms as large as 900,000 Da) located at the M-line.
In vertebrates, sarcomeres are 2.2-2.5 μm long (distance from Z-disk to Z-disk). However, in many invertebrate muscles, sarcomeres are much longer: in adult C. elegans body wall muscle, the sarcomere is about 12 μm long, with thick filaments and A-bands spanning 10 μm. A single polypeptide of vertebrate titin (~3 MDa and 1.2 μm long) spans half a sarcomere, with its C-terminus at the M-line and N-terminus at the Z-disk. However, the dimensions of invertebrate sarcomeres preclude even the possibility of such a polypeptide—it would need to span 6 μm, and have a molecular mass of 15 MDa. Such proteins have not been identified, and not predicted from analysis of genome sequences. Invertebrate titins, such as TTN-1 in C. elegans or D-titin in Drosophila, are ≈2 MDa and are restricted to the I-band. One hypothesis is that the function of one titin in vertebrates is served by one continuous polymer of 3 proteins in invertebrates—UNC-89 at the M-line, twitchin in the A-band (multiple molecules), and TTN-1 in the I-band. We already have a hint of such an overlap for twitchin and TTN-1, although each confocal image is not necessarily consistent with this conclusion (Forbes et al., 2010). Using conventional confocal microscopy, it has not been determined whether twitchin is localized continuously throughout polar regions of the A-band (e.g., as a polymer), or whether it is located periodically with gaps. Hopefully, super-resolution fluorescence microscopy or immuno-Au EM, combined with epitope-specific antibodies, will resolve these questions.
One of the first protein kinases to have its structure elucidated was that of twitchin kinase (Hu et al., 1994). Like the more distantly related myosin light chain kinase, a 60 residue “C-terminal regulatory domain (CRD)”, which lies just C-terminal of the catalytic core of the enzyme, inhibits enzymatic activity (Lei et al., 1994). In vitro, using fragments of twitchin kinase, catalytic activity towards a model peptide substrate is low when the CRD is present, and increased when the CRD is missing. The crystal structure of C. elegans twitchin kinase (kinase-CRD) revealed that the CRD wedges itself between the two lobes of the catalytic core, and blocks the binding sites for ATP and protein substrate. A similar conformation of the CRD and catalytic core was verified with the crystal structures of Aplysia twitchin kinase (Kobe et al., 1996), and human titin kinase (Mayans et al., 1998).
However, how activation of these protein kinases occurs in vivo is unknown. Although calmodulin binds to the CRD of twitchin kinase in vitro, it does not lead to significant activation (Lei et al., 1994). It was hypothesized that activation results from small mechanical pulling forces, occurring during normal muscle activity, that are sufficient to remove the CRD from the catalytic pocket and permit binding to substrates. This model was based on steered molecular dynamics (SMD) simulations on human titin kinase (Grater et al., 2005), and was in agreement with the observed functional association of titin kinase with certain proteins in a stretch-activated manner (Lange et al., 2005). There is also some evidence that stretching force activates twitchin kinase in Mytilus: with permeabilized smooth muscles, a 10% stretch results in a 2-fold increase in phosphorylation of a model substrate for molluscan twitchin kinase in vitro (Butler and Siegman, 2011). Single molecule experiments of titin kinase using atomic force microscopy (AFM) can demonstrate that ATP binds to stretch-induced forms of the kinase (Puchner et al., 2008). However, stretch-induced catalysis has not yet been proven for titin kinase or any other giant kinase. Seeking to obtain experimental evidence for the force-induced unfolding of the CRD suggested by the SMD simulations, AFM was used to pull on single molecules of C. elegans twitchin kinase (Greene et al., 2008). This was the first time that the response of any kinase to pulling force was reported. It revealed that twitchin kinase is significantly resistant to force, unfolding at ≈50-80 pN, and surrounded by Ig and Fn3 domains that are more resistant, unfolding at approximately 100 pN. This is what would be expected if the kinase domains of the giant proteins act as force sensors. Moreover, the AFM data demonstrates that the kinase domains unfold in a step-wise manner (rather than all at once for Ig or Fn3 domains), first with the unfolding of the β-sheet-rich smaller lobe, followed by unfolding of the α-helical larger lobe. The sought-after movement of the CRD from the catalytic pocket was not observed, although the noise of the instrument was probably too high.
The crystal structure, in vitro kinase assays, and SMDs of a larger segment of C. elegans twitchin (Fn-NL-kinase-CRD-Ig), indicate that the regulation of twitchin kinase activity is more complex than previously appreciated (von Castelmur et al., 2012). The structure (Figure 9) revealed for the first time that an N-terminal linker (NL), of 45 residues that lies between the Fn and kinase catalytic core, forms a “crown” that rests on the back of the interlobular kinase hinge region, with its remaining chain folded against the N-terminal lobe, sprawling across the β1-β2 hairpin (containing the glycine-rich loop in the ATP binding pocket). The NL also makes direct contacts with the catalytic helix αC. Kinase assays reveal that both the CRD and the NL each inhibit catalysis by half, and if both are present, kinase activity is totally inhibited. SMD of this larger structure indicates that it is the NL, and not the CRD, that is mechanically sensitive. In fact, during the SMD, even after the N-terminal lobe has unwound, the CRD remains bound to the folded C-terminal kinase lobe. This, together with the observed catalytic tolerance of twitchin kinase to the CRD, suggests that the CRD might help maintain stabilizing contacts with the kinase domain throughout the catalytic cycle, protecting the active site from mechanical damage (von Castelmur et al., 2012). However, whether the CRD is ever removed in vivo, and if so, how it is removed remains a mystery.
|
Figure 9. Crystal structure of a portion of twitchin containing its protein kinase domain. (A) Schematic representation of domains in the originally described twitchin molecule. Ig domains, violet; Fn3 domains, green; protein kinase domain, as indicated; non-domain regions, clear. (B) On top shows a region of twitchin near its C-terminus, and in color, which portions were included in the new crystal structure. The numbering of domains corresponds to the numbering of domains in (A). NL denotes the N-terminal linker, a 45-residue segment lying between Fn31 and the kinase catalytic core (kin); CRD denotes the “C terminal regulatory domain”, a 60-residue segment that lies between the kinase catalytic core and Ig26. Below shows the crystal structure in three rotated views. A white arrowhead indicates helix αR2 of CRD that blocks the ATP binding site. The CRD wedges itself between the two lobes of the catalytic core (shown in grey space-filling mode), and blocks the binding sites for ATP and protein substrate. The NL forms a “crown” that rests on the back of the interlobe kinase hinge region, with its remaining chain folded against the N-terminal lobe, sprawling across the β1-β2 hairpin containing the glycine-rich loop in the ATP binding pocket. The NL also makes direct contacts with the catalytic helix αC. In vitro kinase assays show that both the CRD and the NL each inhibit catalysis by one-half. In steered molecular dynamics (SMD) simulations, it is the NL that first responds to pulling force, whereas the CRD remains bound to the C-terminal kinase lobe. Note that in part B, both in the schematic drawing and crystal structures, the Fn domain is blue and the Ig domain is green (different from Figure 9A and Figure 10). (B, reprinted, with permission, from von Castelmur et al., 2012).
A recent report (Matsunaga et al., 2015) suggests that the binding to and/or phosphorylation of twitchin's CRD by MAPKAP kinase 2 is involved in removing the CRD from twitchin's catalytic pocket. Clones representing a C. elegans ortholog of MAPKAP kinase 2 called MAK-1 were isolated upon screening a Y2H library using the twitchin fragment Ig-Fn-NL-kinase-CRD. Minimally, for this interaction to occur, the NL-kinase-CRD of twitchin and residues 81-405 (essentially 58 residues N-terminal of the kinase domain) of MAK-1 are required. By Y2H assays, MAK-1 interacts with twitchin kinase but not comparable regions of UNC-89 or TTN-1. Analysis of a series of twitchin/TTN-1 chimeras indicated that a 140 aa region containing the end of the kinase catalytic core and most of the CRD is crucial for interaction. Using in vitro binding assays with purified recombinant proteins, the CRD was found to be important for binding to MAK-1. The mak-1 promoter is expressed in body wall muscle, the intestine, and the hypodermis. Antibodies to MAK-1 localize between and around dense bodies, extending into the outer edges of the A-band where there is overlap with twitchin. A likely null allele, mak-1(ok2987), has normal muscle structure but slightly reduced motility in a swimming assay. Exposure of wild type animals to 0.1% nicotine results in paralysis within 30 minutes, but all unc-22 mutants continue to move. mak-1(ok2987) shows partial resistance to nicotine. A mak-1(ok2987); unc-22 (RNAi) double mutant shows complete resistance to nicotine. This epistasis suggests that MAK-1 may phosphorylate twitchin kinase. In in vitro kinase assays, active MAK-1 can phosphorylate catalytically dead twitchin kinase. The following model was proposed: the flanking NL and CRD sequences each inhibit twitchin kinase activity by one half; partial activation occurs by mechanical pulling force to remove the NL from the catalytic pocket; and additional or independent partial activation occurs by the binding and/or phosphorylation of twitchin's CRD by MAK-1. It should be pointed out that physiological substrates for twitchin kinase have not yet been identified.
In C. elegans, the largest polypeptide known is TTN-1 (originally called “CeTitin”), which has a molecular mass of 2.2 MDa. This giant polypeptide was first predicted by analysis of genomic sequence and partial cDNAs (Flaherty et al., 2002), and later detected with antibodies (EU143 and 9/10) to two portions of TTN-1 on Western blot (Forbes et al., 2010). On WormBase, based on predictions and partial cDNAs, 7 isoforms are listed, generated by alternative 3’ ends and alternative splicing. They range in size from ≈18,500 residues for isoforms e, f, and g, to 10,578 residues for isoform d, 3,215 residues for isoform c, and ≈700 residues for isoforms a and b. On the opposite strand of the largest intron for ttn-1 (9.5 kb) resides the mutationally defined gene exp-2, which encodes a K+ channel (Flaherty et al., 2002). By immunofluorescence staining, antibodies to TTN-1 localize to the I-band, and may extend into the outer edge of the A-band (Forbes et al., 2010). Consistent with this localization, six different 300-residue segments of TTN-1 were shown to variously interact with actin and/or myosin in vitro, using F-actin pelleting assays and myosin ELISAs, respectively (Forbes et al., 2010). It should be noted that the first report of TTN-1 localizing between dense bodies (Flaherty et al., 2002)), in retrospect, was an artifact. The antibody, EU102, had been generated to a portion of TTN-1, but later, surprisingly, was found to only react with KETN-1 (kettin), another poly-Ig domain polypeptide of large size (472 kDa) specific for invertebrates. In transgenic animals, the N-terminal 100 residues of TTN-1 tagged with GFP localizes to dense bodies (Flaherty et al., 2002). Therefore, it is likely that single TTN-1 polypeptides have their N-termini embedded at the dense body, and their C-termini extending into the I-band, perhaps into the outer edge of the A-band.
The largest isoform of TTN-1 resembles twitchin in that it contains multiple Ig (56 total) and Fn3 (11 total) domains, and a single protein kinase domain near its C-terminus. In addition, TTN-1 contains five classes of short (14- to 51-residue) repeat motifs arranged mostly as tandem copies: 39- residue repeats forming an ≈2400-residue “PEVT region” similar in amino acid composition to PPAK repeats of the PEVK region, the main elastic region of vertebrate titin; 51-residue CEEEI repeats that interrupt the PEVT repeats, similar to the E-rich repeats that interrupt the PPAK repeats in titin's PEVK region; 14-residue repeats making up the 254-residue AAPLE region; 16-residue “BLUE” repeats that make up an ≈1500-residue region originally predicted to form a coiled-coil structure; and a 30-residue DispRep repeat present in 15 dispersed copies that punctuate other originally predicted coiled-coil regions. Conformations of synthetic peptides of representative copies of each of the five classes of repeats were studied by circular dichroism (Forbes et al., 2010). The data indicate that the PEVT, CEEEI, APPLE, and DispRep regions are all intrinsically disordered and quite similar to the conformational malleability and elasticity of vertebrate titin PEVK segments. Circular dichroism and modeling studies suggest that the BLUE repeats form long, modular, and unstable α-helical oligomerization domains, suggesting that TTN-1 could perhaps bundle.
Like twitchin kinase, TTN-1 kinase has in vitro kinase activity towards a model peptide derived from vertebrate myosin light chains. Intriguingly, alternative splicing results in two isoforms of the CRD (one is 60 residues, the other is 75 residues), and protein kinase domains with these different-length CRDs have different kinase activities (Flaherty et al., 2002). A candidate in vivo substrate for TTN-1 kinase is the dense body UNC-112 interactor UIG-1. The phosyphorylation of UIG-1 by TTN-1 may inhibit interaction with UNC-112 (D. Greene, H. Qadota, and G.M. Benian, unpublished data). Single molecule AFM experiments have revealed that the segment of TTN-1, Fn-kinase-Ig, responds similarly to force as twitchin (Greene et al., 2008). The kinase domain of TTN-1 is most similar to the kinase domains of twitchin (54% identical) and less similar to vertebrate titin kinase (39% identical). Interestingly, there is evidence that vertebrate titin kinase is likely to be an inactive pseudokinase (Bogomolovas et al., 2014). Therefore, C. elegans TTN-1 can be regarded as a “hybrid” between invertebrate twitchin, due to its homologous kinase domain with demonstrable in vitro phosphotransferase activity, and vertebrate titin, due to its multiple tandem repeat regions that are similar to titin PEVK.
Currently, a mutant phenotype for ttn-1 has not been described. Preliminary RNAi experiments using multiple regions of the coding sequence have failed to reveal anything significant (D.B. Flaherty and G.M. Benian, unpublished data). The MMP (Thompson et al., 2013) lists 374 mutations in ttn-1, including 13 nonsense mutations, 3 splicing defects, 2 with deletions in exons, and 2 with insertions leading to frameshifts in exons. Certainly, this issue is ripe for exploration.
In C. elegans, unc-89 loss-of-function mutants display reduced locomotion, disorganized myofibrils, and lack M-lines (Waterston et al., 1980; Benian et al., 1999; Small et al., 2004). unc-89 mutants show disorganization of myosin thick filaments by immunostaining of late larvae and adults (Qadota et al., 2008b; Wilson et al., 2012), although at the L1 larval stage thick filaments are properly organized (Spooner et al., 2012). This indicates that UNC-89 is required either for maintenance of sarcomere organization or growth of sarcomeres. unc-89 is a complex gene: through the use of three promoters and alternative splicing at least eight major polypeptides are generated, ranging in size from 156,000 to 900,000 Da (Benian et al., 1996; Small et al., 2004; Ferrara et al., 2005). The largest of these isoforms, UNC-89-B and UNC-89-F, consist of 53 Ig domains, two Fn3 domains, a triplet of SH3, DH and PH domains near their N-termini, and two protein kinase domains (called PK1 and PK2) near their C termini (Figure 10). Antibodies localize UNC-89 to the M-line (Benian et al., 1996; Small et al., 2004).
|
Figure 10. Progress in identifying proteins that interact with the giant protein UNC-89. On top is a schematic representation of domains in the largest UNC-89 isoform, UNC-89-B, which contains 8081 residues. Ig domains, violet; Fn3 domains, green; a 645 residue long region containing multiple copies of the 3 amino acid triplet KSP, yellow; protein kinase domains, SH3, DH, and PH domains, as indicated. Open rectangles beneath this schematic denote segments used for screening a Y2H library. Interactions have been verified by a combination of demonstrating binding in vitro using purified recombinant proteins, co-immunolocalization in muscle, and identifying muscle mutant phenotypes. Interacting proteins identified so far are indicated as ovals with their protein names (including in parentheses the names of the human homolog or nature of the protein) with arrows leading from the segments of UNC-89 that interact with them. Brief descriptions of the type of protein (e.g., CTD-type phosphatase) and/or mutant phenotype of the interacting protein (e.g., Pat embryonic lethal) are indicated.
The human homolog of UNC-89 is obscurin, which was identified five years after UNC-89 was characterized at the molecular level (Bang et al., 2001; Young et al., 2001). While obscurin contains all the same domains as UNC-89, the SH3, DH, and PH domains are located near the C-terminus rather than near the N-terminus as they are in UNC-89. Although UNC-89 is located only at the M-line, various obscurin isoforms are located at either the M-line, or the Z-disk (Young et al., 2001; Bowman et al., 2007).
To learn how UNC-89 is localized and performs its functions, the Benian laboratory is systematically identifying its binding partners. The coding sequence for the largest isoform, UNC-89-B, is entirely represented as a set of 17 overlapping segments in Y2H vectors. All 17 have been used to screen both a Y2H bookshelf of 23 known components of the nematode M-line, and a Y2H cDNA library. Published interactions are summarized in Figure 10. Each of the kinase domains, PK1 and PK2, interact with SCPL-1 (Qadota et al., 2008a), a CTD-type protein phosphatase. Previously, CTD phosphatases were known to be involved in the regulation of transcription. The UNC-89 to SCPL interaction suggests a new function for this class of phosphatases in sarcomere specific signaling through giant muscle protein kinases. scpl-1 mutants show “hyper-bending” during locomotion (Nahabedian et al., 2012; see above), and are defective in egg laying muscle function (Qadota et al., 2008a). Both the PK1 region and the “interkinase region” each interact with LIM-9 (Xiong et al., 2009), the closest worm homolog of the human protein FHL2 (“four and a half LIM domains” protein) (Lecroisey et al., 2013). lim-9(RNAi) shows aggregates of myosin (Meissner et al., 2009). Recently, a similar interaction between vertebrate obscurin and FHL2 has been reported (Hu and Kontrogianni-Konstantopoulos, 2013). As noted above, LIM-9 was identified as a binding partner of UNC-97 (PINCH) (Qadota et al., 2007), a member of the four protein complex associated with integrin, suggesting a connection between the integrin adhesion complex and UNC-89.
The DH-PH region of UNC-89 activates RHO-1 (RhoA) specifically, and attenuated RNAi for rho-1 results in disorganization of muscle thick filaments (Qadota et al., 2008b). A similar interaction between RhoA and obscurin has also been demonstrated (Ford-Speelman et al., 2009). Ig1-3 of UNC-89 interacts with CPNA-1, a copine domain protein with human and mouse homologs, located at both M-lines and dense bodies (Warner et al., 2013). As noted above, loss-of-function of cpna-1 is Pat embryonic lethal (Meissner et al., 2009). Although CPNA-1 is not required for initial assembly of UNC-89 and MHC A at the M-line, it is required for retention of these proteins at the M-line, once embryonic muscle contraction begins. It is interesting to note that the N-terminal half of PAT-6 (actopaxin) interacts with CPNA-1. This interaction is supported by genetic data: in embryos, in a pat-6 null mutant, CPNA-1 is mis-localized; in adults subjected to pat-6(RNAi), CPNA-1 is found in abnormal accumulations or misclocalized (Warner et al., 2013). The PAT-6 to CPNA-1 to UNC-89 interactions suggests another way in which UNC-89 is localized to the integrin adhesion complex of the M-line.
Most recently, the SH3 domain of UNC-89 has been reported to interact with paramyosin, the invertebrate-specific protein with significant homology with myosin rods (Qadota et al., 2016). The SH3 domain interacts with an 82 amino acid segment (residues 294-376) of the mostly coiled-coil 873 residue-long paramyosin. unc-89 mutants lacking expression of giant isoforms that contain the SH3 domain (e.g., su75 and r452) show paramyosin aggregates. Overexpression of the SH3 domain results in mis-localization of paramyosin. This interaction is unexpected for two reasons. First, SH3 domains usually interact with proline rich sequences that are absent in coiled-coil proteins like paramyosin. Nevertheless, there are several targets of SH3 domains that have been shown to lack the proline-rich consensus sequence (reviewed in Saksela and Permi, 2012). Second, the model for the structure of the nematode thick filament is that it consists of an outer layer of myosins MHC A and B, an intermediate layer of paramyosin, and an inner layer of paramyosin and filagenins (Deitiker and Epstein, 1993; Liu et al., 1998). The vertebrate M-line is observed by EM to be a structure in which the shafts of the thick filaments are crosslinked at their surfaces by a series of struts and additional filaments (Knappeis and Carlsen, 1968; Luther and Squire, 1978). Assuming that the nematode M-line has a similar structure, it is difficult to understand how UNC-89 interacts with the thick filament. One possibility is that the myosin surface of the thick filament shaft has small openings that allow penetration of UNC-89’s SH3 domain into the intermediate or core layers that contain paramyosin. Another possibility is that during sarcomere assembly paramyosin and UNC-89 associate before they are incorporated into thick filaments and M-lines. In support of this idea, during embryonic muscle development paramyosin is detected in 420 minute embryos (Epstein et al., 1993) during which thick filament precursors are being assembled, and this is the same time at which UNC-89 is first detected (Hresko et al., 1994).
At least five isoforms of UNC-89 contain two protein kinase domains of the titin/twitchin family. The importance of the kinase region of UNC-89 is demonstrated by the fact that three mutant alleles of unc-89, tm752, ok1116, and st79, eliminate all detectable expression of the kinase-containing isoforms, but retain the non-kinase containing isoforms (Ferrara et al., 2005), and show disorganization of the myofilament array by polarized light microscopy. UNC-89 isoforms -B, -F, and –H contain two complete protein kinase domains called PK1 and PK2 (Small et al., 2004). In contrast, UNC-89-C and –D, controlled by alternative promoters, begin with partial kinase domains. Homology modeling shows that PK1-C and PK1-D are the large lobes of a protein kinase domain, preceded by a hinge, and homologous N-terminal tails that form similar β-turn-β structures. The functional significance of such partial kinase domains is unknown. Each complete kinase domain found in the other isoforms follows the topology of a typical two-lobed kinase. However, examination of the residues forming an expected ATP binding pocket suggests that PK2 is an active kinase, but that PK1 is not catalytically active. Using in vitro kinase assays, PK2 can phosphorylate several model peptide substrates, and thus, indeed is an active kinase (T. Ferrara and G.M. Benian, unpublished data).
The general structure of a kinase, an intervening sequence of 450-1100 residues, and a second kinase, is conserved in four proteins: C. elegans UNC-89, fly Unc-89 (obscurin), vertebrate obscurin, and SPEGβ. Within the intervening or interkinase region, all four proteins have an Ig followed by an Fn3 domain N-terminal of the second kinase, but otherwise there is no significant homology. The interkinase region has low sequence complexity in all four proteins and high proline content in UNC-89, obscurin, and SPEGβ (11.8, 13.7 and 13.3%). The interkinase regions are similar in length for UNC-89, obscurin, and SPEGβ (905, 955, and 1098 aa), but shorter in fly Unc-89 (457 aa). The low sequence complexity and high proline content is reminiscent of the well-known molecular springs of titin (PEVK, N2B, and N2A). Preliminary experiments using AFM demonstrate that application of force upon the interkinase of UNC-89 results in a smooth and gradual force extension curve consistent with a highly elastic random coil (A.F. Oberhauser and G.M. Benian, unpublished data).
VAV-1 is a guanine nucleotide exchange factor (GEF) for Rac that regulates the concentration of intracellular Ca2+ and rhythmic behaviors such as pharyngeal pumping (Norman et al., 2005). vav-1 is widely expressed, including in the pharynx and body wall muscle. Overexpression of vav-1 in body wall muscle results in decreased locomotion. Mutagenesis of this strain led to isolation of suppressor mutations that reduce impaired movement; these suppressors are loss-of-function alleles of egl-19 and unc-89 (Spooner et al., 2012). EGL-19 is an L-type voltage gated Ca2+ channel, and isolation of an egl-19 mutant is consistent with VAV-1 regulating Ca2+ signaling. However, isolation of an unc-89 mutant, ak155, was unexpected. This finding is compatible with the previously described role of obscurin as a linking molecule between sarcomere and sarcoplasmic reticulum (SR), through interaction of obscurin with the SR membrane protein small ankyrin 1 and 2 (Bagnato et al., 2003; Kontrogianni-Konstantopoulos et al., 2003). It is also compatible with the disorganization of the SR observed in skeletal muscle in the obscurin knockout mouse (Lange et al., 2009).
To obtain more direct evidence that UNC-89 has a role in Ca2+ signaling, a gof mutation in egl-19 that shows a short hyper-contracted body phenotype was used. The double mutant, unc-89(ak155); egl-19(gof) was suppressed for the hyper-contracted phenotype (Spooner et al., 2012). Both the vav-1 overexpression and egl-19(gof) phenotypes can be suppressed by the canonical unc-89 allele, e1460. Both ak155 and e1460 are nonsense mutations lying in the coding sequences for Ig domains 15 and 21, respectively. These mutations are predicted to disrupt the large UNC-89 isoforms, but not the short kinase-containing isoforms, since the short isoforms are controlled by promoters lying downstream of the ak155 and e1460 mutation sites. unc-89(st79), which contains a stop codon in the Ig domain lying just N-terminal of the second kinase domain (Ferrara et al., 2005) and does not express the small isoforms, cannot suppress vav-1(gof) and egl-19(gof) (Spooner et al., 2012). Similarly, RNAi of the kinase containing isoforms of unc-89 could not suppress vav-1(gof), while RNAi of the large UNC-89 isoforms could. Therefore, the large Ig-rich isoforms of UNC-89, but not the kinase encoding isoforms of UNC-89, are involved in Ca2+ signaling. Also, it was found that unc-89 acts in the same genetic pathway as egl-19 and unc-68, which encodes the ryanodine receptor (RyR), and both the RyR and the SR calcium ATPase (SERCA), a Ca2+ reuptake pump, are mislocalized in unc-89 mutants lacking the large Ig-rich isoforms (Spooner et al., 2012). These data suggest that UNC-89 plays a critical role in Ca2+ signaling in the body wall muscle. Indeed, using the Ca2+ sensor, yellow cameleon YC3.60, although there are reliable Ca2+ transients in unc-89(ak155), the peak amplitudes are reduced and there is a delay in the time required to reach maximal Ca2+ concentration, as compared to wild type (Spooner et al., 2012).
The functional implications of the different UNC-89 interactions are various: CPNA-1 stabilizes UNC-89 at the M-line in embryonic muscle, the interaction of UNC-89 with UNC-15 (paramyosin) is important for thick filament assembly or stability, and interaction with LIM-9 helps UNC-89 localize to the M-line through LIM-9’s interaction with UNC-97 and UNC-97’s interaction (via UNC-112, PAT-4 and PAT-6) with integrin. The other interactions seem related to UNC-89’s influence on enzymatic activities at the M-line—thus, the DH-PH region of UNC-89 activates RHO-1 (RhoA), UNC-89 is likely to inhibit the activity of the CUL-3/MEL-26 complex from promoting ubiquitin-mediated degradation of MEI-1 and possibly other substrates, the interaction of UNC-89 kinase domains with SCPL-1 may be involved in some type of signaling in which the kinase activates or inhibits the phosphatase (or vice versa), or each has antagonistic activity towards the same unknown substrate(s).
The skeletal muscle isoform of human dystrophin is a 427 kDa (3685 amino acids) sub-sarcolemmal protein. The X-linked dystrophin gene is 2.5Mb, making it one of the largest human genes, occupying about 1% of the X chromosome. Mutations that abrogate dystrophin function lead to Duchenne Muscular Dystrophy (DMD), a severe progressive muscle wasting pathology, affecting about one out of 3500 male births (Hoffman et al., 1987; Koenig et al., 1987; Emery, 1991; Emery, 2002). Dystrophin localizes at costamere muscle adhesion complexes and the neuromuscular junction (NMJ) (Rybakova et al., 2000; Pilgram et al., 2010). The protein binds on its N-terminal end to the actin cytoskeleton, and on the C-terminal end to a large multi-protein complex, the Dystrophin Glycoprotein Complex (DGC), thus forming a bridge between the cytoskeleton and the sarcolemma and extracellular matrix. The DGC is composed of at least 10 proteins: the intracellular proteins α-dystrobrevin and syntrophins, the trans-membrane proteins β-dystroglycan, α-β-γ-δ- sarcoglycans, and sarcospan, and the extra-cellular proteins laminin 2 and α-dystroglycan (reviewed in Blake et al., 2002).
The highly conserved dystrophin protein consists of three major domains. In the N-terminus the actinin-like domain is important for actin binding. The C-terminal region contains a cysteine-rich domain as well as WW, EF, hand and ZZ modules, which are important for dystrophin to bind to β-dystroglycan, and a coiled-coil domain which is important for dystrobrevin binding. The rod region is composed of 24 helical spectrin-like repeats which are important for actin binding as well as phospholipid binding in addition to structural integrity.
Dystrophin is thought to play a structural role in ensuring membrane stability and force transduction during muscle contraction. In addition, the DGC provides a scaffold for various signaling and channel proteins, which may implicate dystrophin and the DGC in regulation of signaling processes (Constantin, 2014). However, it has not been clearly established yet why muscles degenerate in the absence of dystrophin, and in particular whether the pathology observed in DMD patients is due to a mechanical weakness of the sarcolemma, or if it results from a secondary impairment of proteins displaced because of the absence of dystrophin.
C. elegans has a dystrophin-like protein encoded by the dys-1 gene. The dys-1 gene is expressed in pharyngeal, vulval and body wall muscles of C. elegans (Bessou et al., 1998). Sub-cellular analyses using a monoclonal antibody to DYS-1 or a DYS-1::GFP expressing strain showed that DYS-1 localizes in body wall muscles similarly to its vertebrate counterparts—under the sarcolemma of striated body wall muscle, at I bands near the dense bodies, and in muscle arms (the postsynaptic elements of the NMJ in C. elegans muscle) (Dixon and Roy, 2005; Brouilly et al., 2015).
The dys-1 gene contains 46 exons spanning 31 Kb. DYS-1 has two distinct isoforms, though the function of the short isoform B is currently unknown and will not be further discussed here. Isoform A is a 3,674-amino acids protein (Figure 11), which shares extensive sequence similarities with its mammalian counterparts, including several similar key motifs: the N-terminal actinin-like domain (20% homology with human dystrophin), the central rod-like domain (10 % of homology with human dystrophin) composed of repetitions of spectrin-like triple helices, the C-terminal region (37% of homology with human dystrophin) containing a WW domain, a cysteine-rich domain, and at the very C-terminal end, a coiled-coil domain. Furthermore, the nematode protein seems to have some functional properties in common with its human counterpart, since a chimeric gene containing 80% of human dystrophin coding sequence is able to partly rescue the phenotype of dys-1 mutants (Bessou et al., 1998).
|
Figure 11. Structure of human and C. elegans dystrophin proteins. The structure of the large muscular isoform of human muscular dystrophin is presented (adapted from Blake et al., 2002) along with the two isoforms (A and B) of C. elegans DYS-1. The size of human dystrophin and DYS-1 isoform A is approximately analogous. The N-terminal ends of these proteins share 20% homology and correspond to an actinin/calponin like actin binding domain (blue). The C-terminal ends share 37% homology and contain WW, EF Hand, and Zinc Finger (ZZ) motifs as well as coiled-coil domains (orange). The central rod domains, which shares only 10% homology, contains helical spectrin-like repeats (purple). The distribution and abundance of spectrin repeats varies between species, 24 were found in the sequence of human dystrophin and 7 in DYS-1A. The predicted short isoform DYS-1B corresponds to the last 237 amino acids of isoform A. Figure designed by M. Baritaud.
dys-1 loss-of-function mutants are viable, undergo slight muscle degeneration (see below), and have a peculiar phenotype consisting of hyperactivity, exaggerated head bending, and a tendency to hyper-contract. Moreover, dys-1 mutants are hypersensitive to acetylcholine and to the acetylcholine esterase (AchE) inhibitor aldicarb, suggesting that the absence of functional dystrophin up-regulates cholinergic transmission in C. elegans (Bessou et al., 1998). It was also shown that AchE activity was reduced in dys-1 mutants (Giugia et al., 1999). However, since dys-1 and AchE mutants have different phenotypes in C. elegans, AchE activity modulation alone was not sufficient to explain the dys-1 mutant phenotype. Evidence for a role of dystrophin in cholinergic transmission came from forward genetic screens aimed at recovering other mutations leading to a dys-1-like phenotype. This work led to the identification of several other genes. One of these genes turned out to encode a dystrobrevin ortholog, a protein known to interact with dystrophin in mammals. Two other genes encoded SLO-1, a calcium-dependent BK potassium channel (Wang et al., 2001), and DYC-1, a protein homologous to the CAPON adaptor protein (Gieseler et al., 2000; Carre-Pierrat et al., 2006; Lecroisey et al., 2008). Finally, the isolation of mutations in the snf-6 gene, leading to the same phenotype as mutations in dys-1, strongly suggested that increased Ach levels at the NMJ may contribute to the dys-1 phenotype (Kim et al., 2004; Ségalat and Anderson, 2005). Indeed, the snf-6 gene encodes an acetylcholine/choline transporter, which regulates the uptake of acetylcholine at neuromuscular junctions.
In addition to dystrophin (DYS-1) and dystrobrevin (DYB-1), C. elegans has other orthologs of vertebrate DGC proteins, such as δ/γ-sarcoglycan (SGN-1), syntrophins (STN-1 and -2), α- and β-sarcoglycans (SGCA-1 and SGCB-1), and dystroglycan (DGN-1). No sarcospan counterpart was found (Grisoni et al., 2002; our unpublished results). DYS-1 was shown to interact with DYB-1 and STN-1 suggesting that a DGC may exist in C. elegans (Gieseler et al., 1999a; Gieseler et al., 1999b; Grisoni et al., 2003). In addition, mutation or RNAi mediated gene knock down led to a dys-1 phenotype for some of the DGC ortholog encoding genes: dyb-1, stn-1, stn-2, sgn-1, and sgca-1 (Gieseler et al., 1999a; Grisoni et al., 2002; Grisoni et al., 2003; Zhou et al., 2008; our unpublished observations). Mutations in the dgn-1 gene lead to different phenotypes and the gene is not expressed in muscle (Johnson et al., 2006), thus dgn-1 function is not linked to dys-1. DYB-1, STN-1, and STN-2, however, are expressed in neurons and striated body wall muscles where all localize at sarcomeres, close to dense bodies. For sgn-1, sgca-1, and sgcb-1 the expression pattern has, to our knowledge, not yet been analysed. Interestingly, STN-1 interacts with SNF-6, and in dys-1 and stn-1 mutants SNF-6 localization at NMJ is lost (Kim et al., 2004). Together, this strongly suggests a role for DYS-1 and the associated DGC in cholinergic neurotransmission at the NMJ, and this role may in part be responsible for the hyperactivity/hypercontraction phenotype of dys-1 and DGC mutants (Figure 12).
|
Figure 12. Functions of DYS-1 and associated proteins in muscle. Part of a muscle cell is shown with one dense body anchored to the distal sarcolemma. Actin containing filaments are anchored to the dense body and interact with myosin containing thick filaments. Cellular organelles such as the nucleus, mitochondria, and endoplasmic reticulum with ribosomes are localized in the deeper part of the muscle cell. A muscle arm corresponding to the postsynaptic elements of the neuromuscular junction in the C. elegans muscle is represented in close contact with a motoneuron. DYS-1 and the associated Dystrophin Glycoprotein Complex (DGC, represented in red) fulfill different functions in muscle. DYS-1 is involved in maintaining dense body and sarcolemma integrtity. DYS-1 localizes under the entire sarcolemma of striated body wall muscle and in muscle arms. Through the interaction of its C-terminal end with the dense body protein DEB-1 (vinculin; blue), DYS-1 links the DGC to the dense body. The LIM-domain protein ZYX-1, which interacts with DEB-1, ATN-1, and DYC-1 may be involved in signaling from the dense body to the nucleus, thus linking DYS-1 and DGC function to dense body signaling. The interaction of ISLO-1 with STN-1 and SLO-1 connects the K+ channel, SLO-1, to the DGC. SLO-1 localizes close to the L-type Ca2+ channel EGL-19. SLO-1 mediated potassium efflux is thought to negatively regulate the calcium-mediated activation of muscle, thus involving DYS-1 and DGC in the control of calcium homeostasis. At the neuromuscular junction acetylcholine (Ach) binding to its receptor (AchR) lead to muscle excitation. DYS-1 and the DGC are involved in cholinergic transmission by controlling ACE-1/2 acetylcholine esterase (AchE) activity and Acetylcholine (Ach) uptake through the acetylcholine/choline transporter SNF-6, which interacts with STN-1.
In addition, several lines of evidence indicate that disturbed calcium homeostasis may contribute to this phenotype. The identification of islo-1, encoding a sub-unit of SLO-1, a voltage-, and calcium-dependent potassium (BK) channel, and the interaction of ISLO-1 with STN-1 and SLO-1 connect SLO-1 to the DGC. Mutations in either slo-1 or islo-1 mimic the dys-1 phenotype. In addition, SLO-1 localizes close to the L-type calcium channel EGL-19, and this localization is lost in either dys-1 or islo-1 mutants. It has, therefore, been proposed that loss of DYS-1, and other DGC components, may contribute to increased calcium-mediated responses by disturbing the SLO-1 mediated potassium efflux thought to negatively regulate the calcium-mediated activation of muscle (Kim et al., 2009). Along this line, another study using in vivo single-molecule imaging identified altered dynamics of the subunit UNC-36 of the L-type calcium channel EGL-19 in dys-1 mutants (Zhan et al., 2014).
In C. elegans the loss of dys-1 function only leads to moderate muscle cell death as observed by progressive loss of actin filaments and muscle nuclei (Bessou et al., 1998; Oh and Kim, 2013). This indicates that the C. elegans muscle is more resistant to the loss of dystrophin function than human muscle. However, the amount of muscle cell death (referred hereafter as muscle degeneration) significantly increased when a dys-1 loss-of-function mutation was combined with a hypomorphic mutation in the hlh-1 gene (Gieseler et al., 2000). In addition, muscle degeneration increases in both the dys-1(cx18) single mutant and the dys-1(cx18); hlh-1(cc561) double mutant with higher body bending forces on firm culture medium, supporting the hypothesis of a mechanical role for DYS-1 (Brouilly et al., 2015). This result is in agreement with studies performed in dystrophin deficient mdx mice that revealed a higher vulnerability and enhanced dystrophic progression in response to exercise (Brussee et al., 1997; De Luca et al., 2003).
Dystrophin plays an important role in linking the cytoskeleton to the sarcolemma and was thus hypothesized to protect against contraction-induced muscle damage via tension transmission and stabilization of the sarcolemma (Blake et al., 2002). In vertebrate skeletal muscle, dystrophin is associated with costameres, which link the sarcomeres to the sarcolemma through an association with Z-disks. The C. elegans dense body fulfills the function of both vertebrate Z-disks and costameres and plays a role in transducing the force generated by sarcomeres to the extracellular matrix (Figure 1A and B; Figure 5). In Y2H experiments, DYS-1 interacts with the vinculin ortholog DEB-1, which localizes at the bases of dense bodies (Brouilly et al., 2015). This suggests that DYS-1 links the DGC to the dense body. Indeed, different proteins belonging to or associated with the DGC have been localized to dense bodies, including DYC-1, which is not directly associated with the DGC but interacts with the LIM domain protein ZYX-1 (ZYX-1 interacts with ATN-1 and DEB-1), and ZYX-1 was shown to be involved in DYS-1 dependent muscle degeneration (Lecroisey et al., 2008; Lecroisey et al., 2013; our unpublished results) (Figure 12). Most interestingly, mutations in DGC ortholog encoding genes not only lead to the dys-1 hyperactivity/hypercontraction phenotype, but also cause muscle degeneration in the sensitized genetic hlh-1(cc561) background: snf-6, slo-1, sgca-1, dyb-1, stn-1, or dyc-1 (Gieseler et al., 1999a; Gieseler et al., 2000; Grisoni et al., 2003; Kim et al., 2004; Carre-Pierrat et al., 2006; Lecroisey et al., 2008; our unpublished results). Along this line it is noteworthy that mutants that exhibit a strong hypercontraction phenotype do not present any muscle degeneration. This is the case for worms with lof mutations in the unc-105 gene, which encodes a member of the Degenerin protein family in C. elegans, involved in mechanical signal transduction and having similarities to the mammalian amiloride-sensitive epithelial sodium channel (ENaC) (Liu et al., 1996). The strong hypercontraction of unc-105 mutants leads to severely impaired movement (Park and Horvitz, 1986) thus preventing muscle degeneration.
The contribution of mechanical forces and movement to DYS-1 dependent muscle degeneration is further supported by a reduction of muscle degeneration in adult dys-1; hlh-1 double mutants when sarcomere contraction is inhibited by mutations in different genes affecting the myofilament lattice (Mariol et al., 2007), RNAi mediated knock down of the EGL-19 L-type voltage gated Ca2+ channel (Mariol and Ségalat 2001), or after forced immobilization by treatment with muscimol, a GABAA agonist and anesthetic used to paralyze C. elegans (Brouilly et al., 2015). Similarly, limb immobilization or treatment with muscle relaxants prevent and reduce the occurrence of muscle degeneration in mdx mice (Mokhtarian et al., 1999). One possible explanation for this observation is that reduced movement and force production allows for the cell to repair damage at dense bodies and plasma membrane. This hypothesis may also explain why most muscle cells degenerate only after the last (L4) larval stage. Indeed, in C. elegans, the cuticle is remade at the end of each of the four larval stages during episodes of physiological lethargic periods (lethargus) (Raizen et al., 2008). Interestingly, the composition and dynamics of the dense bodies change during this quiescent period (Zaidel-Bar et al., 2010), and some dense body proteins are differentially expressed during lethargus (Turek and Bringmann, 2014). In dystrophin-deficient muscle, these periods of quiescence may be used to repair cell damage and the dystrophic phenotype would be consequently delayed and visible only in adulthood when such phases are absent. Alternatively, adult tissues may in general have reduced ability to repair cell damage compared to larvae.
An ultra-structural analysis performed on muscle cells during development, from L1 larvae to 3-days post-L4 dys-1(cx18); hlh-1(cc561) mutants allowed for the observation of the subcellular events occurring during muscle degeneration and to determine their chronology in the degenerative process (Brouilly et al., 2015). The small size of C. elegans makes possible the use of High Pressure Freezing on the whole organism thus reducing fixation artifacts. In addition, C. elegans muscles do not contain satellite cells and are thus not able to regenerate after damage. This model is, therefore, best suited for such a time course study of the subcellular degenerative process. Contrary to degenerative muscle tissues in vertebrates, which present a high degree of spatio-temporal heterogeneity, macrophage infiltration and fibrosis, and uncertain sectioning, using C. elegans reduces heterogeneity and allows a quantitative time-course analysis of muscle degeneration at the ultra-structural level. In this study, the first defects were observed at the very beginning of larval development (L1) with a disorganization of the sarcomeric bundles, presenting an irregular shape when compared to wild type. From the L2 stage, dense body sections of both dys-1(cx18) single and dys-1(cx18); hlh-1(cc561) double mutants were smaller than wild-type. Thus the irregular shape of the sarcomeric bundles could result from organization defects of dense bodies lacking DYS-1. In addition, smaller dense bodies were observed in dys-1(cx18); hlh-1(cc561) mutants at the L3 larval stage and their fragmentation or their detachment from the sarcolemma was frequent (Brouilly et al., 2015; http://www.inmg.fr/en/gieseler/wormboook_gieseler.php). This may explain the disrupted actin network phenotype observed under light microscopy in phalloidin-rhodamine and LifeAct staining experiments. These revealed accumulation of actin in coils and at the tips of the cell, which is probably a consequence of the tension exerted by the excitation/relaxation cycles on dense bodies weakened by the absence of DYS-1, fragmented and detached from the sarcolemma (Gieseler et al., 2000). These observations are in agreement with a role for DYS-1 in the maintenance of dense body anchorage and integrity (Figure 12).
A role for DYS-1 in the maintenance of sarcolemma integrity is suggested by plasma membrane interruptions and a significant increase of single- and double membrane vesicles in dys-1; hlh-1 mutants that were observed by ultra-structural analysis starting from the L3 larval stage. The single membrane vesicles were often located at sub-sarcolemmal regions, suggesting a possible link with plasma membrane repair mechanisms. These mechanisms often involve endo/exocytosis flux in plasma membrane healing or budding (Brouilly et al., 2015).
The disruption of dense body and sarcolemma integrity occurs early in development of dys-1; hlh-1 mutants. In addition to the above-mentioned defects at the neuromuscular junction and the control of ion channel activity, these defects are likely to increase the disruption of cellular homeostasis and to trigger downstream cellular responses. High calcium concentrations were previously observed in muscle of DMD patients and mdx mice (Turner et al., 1988). A calcium overload is also a possible origin for mitochondria dysfunction and mitochondria dynamics, which were observed during muscle degeneration in C. elegans and the zebrafish (Giacomotto et al., 2013). Another consequence of the loss of sarcolemma integrity could be the disturbance of protein homeostasis. Indeed, an acceleration of age-dependent protein aggregation was observed in dys-1 mutant muscle cells (Oh and Kim, 2013). An accumulation of double-membrane vesicles observed in dys-1; hlh-1 mutants starting from the L3 stage could be reminiscent of the activation of autophagy, which participates in the degradation of protein aggregates. Autophagy could further be implicated in the degradation of disrupted muscle filaments and dense bodies, mitochondria as well as ribosomes, and endoplasmic reticulum that were identified within these vesicles (Brouilly et al., 2015; our unpublished results).
Most interestingly, ultra-structural analysis of skeletal muscle biopsies from young DMD patients revealed similar defects to those described in C. elegans dys-1; hlh-1 mutants, such as disruption of Z-disks and sarcomeric bundles, loss of plasma membrane integrity, accumulation of vesicles near the sarcolemma and in internal regions, and abnormal morphology of mitochondria and endoplasmic reticulum (Brouilly et al., 2015). This result emphasizes the suitability of the C. elegans model to unravel mechanisms leading to muscle degeneration diseases in humans. It is likely that in human patients, as in C. elegans, the loss of dystrophin leads to defects in Z-disc and costamere organization, the submembrane cytoskeleton, thus affecting sarcomere attachment to the sarcolemma and membrane integrity. This together with impaired function of NMJ and ion channels could then trigger downstream responses such as membrane repair and protein degradation, which may however not be sufficient to repair cellular damages and to prevent cell death.
Finally, it has to be pointed out that dystrophin-dependent muscle degeneration shares some subcellular features of age-dependent muscle decline, also known as sarcopenia. While sarcopenia has not yet been studied in C. elegans, loss of sarcomere integrity, accumulation of autophagosomes and protein aggregates, and disturbed mitochondrial dynamics and functions have been observed in muscles of aged nematodes that likely contribute to the progressive loss of motility and muscle strength (Herndon et al., 2002; Morley et al., 2002; Yu et al., 2012; Regmi et al., 2014; C. Scholtes and K. Gieseler, unpublished results). Sarcopenia is a long-lasting partially reversible process, which is often qualified as physiologic muscle degeneration in contrast to pathologic muscle degeneration occurring in some neuromuscular disorders such as muscular dystrophies. Pathologic muscle degeneration progresses fast and irreversibly leads to premature muscle cell death. Thus, dystrophin-dependent muscle degeneration may in some aspects be considered as accelerated muscle aging in which additional cellular processes occur that lead to progressive cell death. The precise mechanisms involved in pathologic muscle cell death still need to be clarified (reviewed in Carmelli et al., 2015).
Mice lacking both dystrophin and the myogenic factor MyoD display severe myopathy, compared with mdx mice (Megeney et al., 1996). This increased severity of muscle degeneration in MyoD; mdx double-mutants is thought to be caused by a reduction of muscle regeneration from satellite cells, which requires MyoD function. C. elegans muscle differ from mammalian muscle in that they do not contain satellite cells. Therefore, damaged muscle cells are not replaced in C. elegans. Nevertheless, the hlh-1(cc561) allele of the C. elegans MyoD ortholog potentiates muscle degeneration of dys-1/dystrophin loss-of function mutations.
The hlh-1(cc561) allele is partially thermosensitive and produces a truncated protein, which contains the N-terminal region of HLH-1 including the bHLH domain. At permissive temperature (15 °C), about 60% of eggs have normal development and reach adulthood. Young animals move well and have normal body shape, but contain extra sex muscles (Harfe et al., 1998). At the restrictive temperature (25 °C), severe defects in motility and body shape were observed, with most animals dying as newly hatched larvae (Harfe et al., 1998). The phenotypic effects seen in hlh-1(cc561) animals are likely due to insufficiency rather than novel effects of this truncated protein since the mutant phenotype can be rescued by extra-copies of hlh-1(cc561) (Harfe et al., 1998).
Transgenic animals carrying the GFP sequence fused to the endogenous hlh-1 wild-type or cc561 allele (lines generated by the CRISPR technology), grown at 15 °C, exhibited a GFP signal in all body wall muscle nuclei in larvae and adults. However, the signal was much weaker in hlh-1 (cc561)::gfp animals than in hlh-1(wt)::gfp (E. Martin and K. Gieseler, unpublished results). This observation suggests that the truncated protein is correctly addressed to the nucleus but is less stable than the wild-type form, even at 15 °C.
An ultra-structural analysis performed by transmission electron microscopy at larval stages (L1 to L4) and in young adults revealed that hlh-1(cc561) mutants grown at the permissive temperature have a globally normal body wall muscle structure (Brouilly et al., 2015). However, when combined with a loss-of-function allele of dys-1, the hlh-1(cc561) allele leads, at the permissive temperature for hlh-1(cc561), to a strong progessive muscle degeneration phenotype starting at the L4 and paralysis of adult worms at 3 days post-L4 (Gieseler et al., 2000; Brouilly et al., 2015; see above). This phenotype can also be rescued by extra-copies of hlh-1(wt) or hlh-1(cc561) (our unpublished results), suggesting that the enhancement of muscle degeneration is due to insufficient function of the HLH-1(cc561) protein.
Transcriptomic data obtained on hlh-1(cc561) L4 larvae grown at permissive temperature revealed altered expression of numerous genes that encode proteins involved in lysosomal function, protein turnover and metabolism, or sarcomere and DGC organization (A. Rea-Boutrois, A.R. Reedy, and K. Gieseler, unpublished results). This observation suggests that HLH-1 contributes post-embryonically to cellular homeostasis, sarcomeric protein turnover, and muscle maintenance. While the perturbation of these processes is not sufficient to induce a mutant phenotype in hlh-1(cc561) single mutants, it likely sensitized body wall muscle to other perturbations, thus contributing to the severe muscle degeneration phenotype observed in dys-1(cx18); hlh-1(cc561) double mutants. This hypothesis is further supported by the above mentioned muscle degeneration phenotype observed when the hlh-1(cc561) allele is combined with loss-of-function mutations in either snf-6, slo-1, sgca-1, dyb-1, stn-1, or dyc-11 (Gieseler et al., 1999a; Gieseler et al., 2000; Grisoni et al., 2003; Kim et al., 2004; Carre-Pierrat et al., 2006; Lecroisey et al., 2008; our unpublished results). These mutations all lead to a dys-1-like hyperactivity/hypercontraction phenotype, which is likely to be associated with increased cholinergic transmission and/or disturbed calcium homeostasis. Finally, hlh-1(cc561) mutants also exhibit increased sensitivity of post-embryonic and adult muscle to impaired proteostasis (A.R. Reedy and K. Gieseler, unpublished results), and reduced resistance of muscle during aging (M. Cortes and K. Gieseler, unpublished results).
Altogether, this indicates that in C. elegans HLH-1 may contribute to the repair of cellular damages and prevent cell death. Further research will be required to decipher in detail the role of the myogenic HLH-1 transcription factor in differentiated muscle cells.
Sarcomere assembly and/or maintenance in C. elegans involves at least several hundred different proteins, most of which are conserved in all animals. However, exactly how each of them is involved in assembly and maintenance and how they work together is still largely unknown. Outstanding questions include: how do thick and thin filaments attain fixed and uniform lengths, what determines and maintains the exact spacing and patterning of muscle attachment structures, what determines whether a dense body or an M-line is formed when each begins with the same base proteins, what determines the height and width of dense bodies and M-lines, how are protein components of sarcomeres and other cellular structures repaired and replaced during the stress of muscle activity, what are the essential mechanisms by which loss of dystrophin results in muscle degeneration, what are the targets of the MRFs, what are the full set of pathways that the muscle cell uses to sense its activity, and why are there so many different components of attachment structures that seem not to be functionally redundant? Functional analysis using forward and reverse genetics, cell biology, and biochemistry, especially on the many new components of muscle identified in the last 10-15 years, holds much promise for addressing these and other questions.
Table 1. Notable discoveries about muscle from C. elegans research
| Protein | Contribution | References |
|---|---|---|
| UNC-54 | Cloning, sequencing, and analysis of the first complete myosin heavy chain, UNC-54; model for parallel assembly of myosin rods in the thick filament | Epstein et al. 1974; McLachlan and Karn, 1982; Dibb et al. 1985 |
| DEB-1, UNC-52, PAT-3, etc. | Discovery that perlecan, integrin, and its associated proteins are critical for sarcomere assembly | Barstead and Waterston, 1991; Rogalski et al. 1993; Williams and Waterston, 1994; Hresko et al. 1994; Gettner et al., 1995; Hobert et al., 1999; Rogalski et al. 2000; Mackinnon et al. 2002; Lin et al. 2003 |
| UNC-45 | The first myosin head chaperone, UNC-45 | Epstein and Thomson, 1974; Venolia and Waterston, 1990; Barral et al. 1998; Barral et al. 2002 |
| UNC-22/twitchin | The first complete sequence of a giant titin-like protein twitchin; the first intracellular protein to join the Ig superfamily | Moerman et al. 1986; Moerman et al. 1988; Benian et al. 1989; Benian et al. 1993 |
| UNC-22/twitchin | The first crystal structure of a protein kinase domain from a titin family member, that of twitchin kinase, and structural basis of autoinhibition | Hu et al. 1994; von Castelmur et al. 2012 |
| UNC-89 | Function and domain organization of the first obscurin family member, UNC-89 | Waterston et al. 1980; Benian et al. 1996; Small et al. 2004 |
| UNC-112 | First recognition that kindlins have a role in integrin adhesion complexes by cloning and mutational analysis of UNC-112 | Rogalski et al. 2000 |
| UNC-60 | First recognition that actin regulatory proteins are crucial for sarcomere assembly or maintenance by the cloning, mutational, and biochemical analysis of UNC-60 (ADF/cofilin) | Waterston et al. 1980; McKim et al. 1994; Ono et al. 1999 |
| HSP-1, UNC-23 | First recognition that a molecular chaperone, HSP-1 (Hsc70), and its regulators (including UNC-23 (BAG-2)) are required to maintain muscle adhesion complexes in the face of mechanical stress | Waterston et al. 1980; Plenefisch et al. 2000; Papsdorf et al. 2014; Rahmani et al. 2015 |
| DYS-1 | First ultrastructural chronology of dystrophin-dependent muscle degeneration | Brouilly et al. 2015 |
Table 2. Proteins that function in muscle assembly, maintenance or function*
| Protein | Human homolog/ortholog | Location | Interacting partners | Phenotype of mutants | Key references |
|---|---|---|---|---|---|
| ALP-1 | cypher/ZASP/oracle | dense body (deeper part), nucleus (muscle and hypodermis) | ZYX-1, ATN-1? | normal thrashing assay; reduced maximal bending; low penetrance F-actin aggregates enhanced by atn-1/+ or ketn-1(RNAi) | McKeown et al. 2006; Han and Beckerle 2009 |
| ATN-1 | α-actinin | dense body (deeper part) | DEB-1, ZYX-1, ALP-1? | short and broad dense bodies; F-actin aggregates; reduced maximum bending | Barstead et al. 1991; Moulder et al. 2010 |
| CPNA-1 | CPNE5 | M-line, dense body | PAT-6, UNC-89, LIM-9, UNC-96, SCPL-1 | Pat, at 2 fold stage UNC-89 and MYO-3 form accumulations | Warner et al. 2013 |
| CSN-5 | CSN5 | A-band | UNC-96, UNC-98 | RNAi increases steady state level of UNC-98 | Miller et al. 2009 |
| CYK-1 | formin | dense body | narrow muscle cells, fewer striations per cell | Mi-Mi et al. 2012 | |
| DAF-21 | Hsp90 | transiently at and diffusable from I-band | UNC-45, UNC-54? | reduced motility in liquid; aggregates of GFP::MYO-3 outside myofilament lattice | Gaiser et al. 2011 |
| DEB-1 | vinculin | dense body (base) | TLN-1, UIG-1, DYS-1, PXL-1, ZYX-1, ATN-1 | Pat | Barstead and Waterston 1989; Barstead and Waterston 1991 |
| DIM-1 | around and between dense bodies | UNC-112, UIG-1 | slight disorganization of myofilament lattice; reduced maximum bending; suppresses unc-112 | Rogalski et al. 2003 | |
| DYB-1 | dystrobrevin | close to dense bodies | DYS-1, STN-1 | hyperactive, hypercontracted, headbending, muscle degeneration in combination with hlh-1(cc561) | Gieseler et al. 1999a; Gieseler et al. 1999b |
| DYC-1 | capon | close to dense bodies | ZYX-1 | hyperactive, hypercontracted, headbending, muscle degeneration in combination with hlh-1(cc561) | Gieseler et al. 2000; Lecroisey et al. 2008 |
| DYS-1 | dystrophin | I-band, dense body, sarcolemma | DYS-1, DYB-1, DEB-1 | hyperactive, hypercontracted, headbending, muscle degeneration in combination with hlh-1(cc561) | Bessou et al. 1998; Brouilly et al. 2015 |
| FHOD-1 | formin | around and between dense bodies | narrow muscle cells, fewer striations per cell | Mi-Mi et al. 2012 | |
| α-filagenin | A-band (center; thick filament cores) | UNC-15?, MYO-3?, UNC-54? | unknown | Liu et al. 2000 | |
| β-filagenin | A-band (polar and center; thick filament cores) | UNC-15?, MYO-3?, UNC-54? | unknown | Liu et al. 1998 | |
| γ-filagenin | A-band (polar regions; thick filament cores) | UNC-15?, MYO-3?, UNC-54? | unknown | Liu et al. 2000 | |
| FRG-1 | FRG1 | dense body, nucleus (nucleoli) | F-actin | overexpression: disruption of some muscle cell-cell junctions and absence of some muscle cells | Liu et al. 2010 |
| HLH-1 | MyoD | nuclei | L1 arrest, paralysis | Krause et al. 1990 | |
| HND-1 | nuclei? | intragenic deletion is embryonic lethal; point mutation is Unc and small fraction are Pat | Baugh et al. 2005 | ||
| HSP-25 | αβ-crystallin | M-line, dense body | DEB-1?, ATN-1? | not fully characterized | Ding and Candido 2000 |
| KETN-1 (kettin) | between dense bodies | F-actin, ATN-1? | by RNAi, F-actin aggregates enhanced by tetramisole-induced hypercontraction and by atn-1 | Ono et al. 2006 | |
| LIM-8 | M-line, around and between dense bodies | UNC-97, UNC-95, PXL-1, ZYX-1, UNC-96, MYO-3, MEL-26 | normal; mildly disorganized MYO-3 in pxl-1; lim-8 double | Qadota et al. 2007; Warner et al. 2011 | |
| LIM-9 | FHL-2 | M-line, around and between dense bodies | UNC-97, UNC-89, ZYX-1, UNC-96, SCPL-1 | mildly disorganized MYO-3 | Qadota et al. 2007; Meissner et al. 2009 |
| MAK-1 | MAPKAP2 | around and between dense bodies | UNC-22 | partially resistant to nicotine; unc-22 is epistatic to mak-1 in nicotine resistance | Matsunaga et al. 2015 |
| MEL-26 | M-line, I-band | CUL-3, MEI-1, UNC-89 | disorganized MYO-3 | Wilson et al. 2012 | |
| MYO-3 (MHC A) | myosin heavy chain | A-band (center) | UNC-98, UNC-96, LIM-8, UNC-15?, UNC-54?, UNC-45? | Pat, severe, very few or no thick filaments | Miller et al. 1983; Miller et al. 1986; Ardizzi and Epstein 1987; Waterston 1989 |
| PAL-1 | nuclei of C and D blastomeres in embryos | embryonic lethal | Hunter and Kenyon 1996; Bowerman et al. 1997; Baugh et al. 2005 | ||
| PAT-2 | integrin α | M-line, dense body (cell membrane) | PAT-3? | Pat; in Pat embryos actin and myosin not polarized | Williams and Waterston 1994 |
| PAT-3 | integrin β | M-line, dense body (cell membrane) | UNC-112, PAT-2? | Pat; in Pat embryos actin and myosin not polarized | Gettner et al. 1995 |
| PAT-4 | integrin linked kinase | M-line, dense body (base) | UNC-112, PAT-6, UNC-97 | Pat; in Pat embryos actin and myosin are polarized, but no thick or thin filaments | Mackinnon et al. 2002 |
| PAT-6 | actopaxin(α-parvin) | M-line, dense body (base) | PAT-4, CPNA-1 | Pat; in Pat embryos actin and myosin are polarized, but no thick or thin filaments | Lin et al. 2003 |
| PAT-9 | nucleus | Pat | Liu et al. 2012 | ||
| PFN-3 | profilin | dense body (deeper part) | F-actin? | slightly slower thrashing assay; reduced maximal bending; null–normal I-bands; F-actin aggregates in pfn-3; unc-94(RNAi) | Polet et al. 2006; Yamashiro et al. 2008 |
| PKN-1 | protein kinase N | M-line, dense body | RHO-1, CED-10, MIG-2 | loopy Unc | Qadota et al. 2011 |
| PXL-1 | paxillin | M-line, dense body | UNC-95, UIG-1, DEB-1, UNC-96, LIM-8 | normal; mildly disorganized MYO-3 in pxl-1; lim-8 double | Warner et al. 2011 |
| RNF-5 | dense body (base) | UNC-95 | dense bodies (stained with anti-DEB-1) not organized in straight rows; overexpression leads to reduced UNC-95; RNAi leads to increased UNC-95 | Didier et al. 2003; Broday et al. 2004 | |
| SCPL-1 | M-line, I-band | UNC-89, CPNA-1, LIM-9, ZYX-1 | increased maximal bending | Qadota et al. 2008a; Nahabedian et al. 2012 | |
| SGCA-1 | muscle degeneration in combination with hlh-1(cc561) | K. Gieseler, unpublished results | |||
| SGCB-1 | K. Gieseler, unpublished results | ||||
| SGN-1 | hyperactive, hypercontracted, headbending | Grisoni et al. 2002 | |||
| STN-1 | close to dense bodies | DYS-1, DYB-1, SNF-6 | hyperactive, hypercontracted, headbending | Grisoni et al. 2003; Kim et al. 2004 | |
| STN-2 | close to dense bodies | SAX-7 | hyperactive, hypercontracted, headbending | Zhou et al. 2008 | |
| TTN-1 | I-band | UIG-1 | unknown | Flaherty et al. 2002; Forbes et al. 2010 | |
| UIG-1 | dense body | UNC-112, CDC-42, DIM-1, TTN-1, TLN-1, DEB-1, PXL-1, UNC-95, ZYX-1 | disorganized myofilament lattice | Hikita et al. 2005 | |
| UNC-15 (paramyosin) | A-band | UNC-54?, MYO-3?,UNC-96, UNC-98, UNC-89 | Unc, limp paralyzed, myofilament lattice very disorganized, shorter hollow thick filaments | Waterston et al. 1977; MacKenzie and Epstein 1980; Kagawa et al. 1989 | |
| UNC-22 (twitchin) | A-band (polar regions) | MAK-1 | Unc, twitching; resistant to nicotine | Moerman et al. 1988; Benian et al. 1989; Hu et al. 1994 | |
| UNC-23 | BAG-2 | M-line, dense body (base), hypodermal cell (nucleus, Ifs) | HSP-1 | Unc, bent, detachment of muscle cells in “head” region | Rahmani et al. 2015 |
| UNC-35 (TLN-1) | talin | dense body, (M-line) | PAT-3?, DEB-1?, F-actin? | Unc, not fully characterized; requires pat-3 but not deb-1 for localization | Moulder et al. 1996; Etheridge et al. 2012 |
| UNC-45 | UNC-45 | A-band (polar regions) | NMY-2, HUM-2, DAF-21, UNC-54? | Pat, Unc; ts allele: reduced number of thick filaments | Venolia and Waterston 1990; Barral et al. 1998; Ao and Pilgrim, 2000; Barral et al. 2002; Kachur et al. 2004 |
| UNC-52 | perlecan | M-line, dense body (ECM) | PAT-2?, PAT-3? | Unc, Pat; in Pat embryos actin and myosin not polarized | Rogalski et al. 1993; Rogalski et al. 1995 |
| UNC-54 (MHC B) | myosin heavy chain | A-band (polar regions) | UNC-22?, UNC-15?, MYO-3?, UNC-45? | Unc, limp paralyzed, myofilament lattice very disorganized, reduced numbers of thick filaments | Epstein et al. 1974; Miller et al. 1983; Karn et al. 1983; Dibb et al. 1985 ; Ardizzi and Epstein 1987 |
| UNC-82 | M-line | Unc, mislocalization of thick filament/M-line proteins during embryonic elongation | Hoppe et al. 2010 | ||
| UNC-89 | obscurin | M-line (full depth) | RHO-1, UNC-15, CPNA-1, MEL-26, LIM-9, SCPL-1 | Unc, missing M-line | Benian et al. 1996; Small et al. 2004 |
| UNC-95 | M-line, dense body, (nucleus?) | RNF-5, UNC-97, PXL-1, LIM-8, UIG-1 | Unc, shorter or broken dense bodies and M-lines | Broday et al. 2004 | |
| UNC-96 | M-line, (dense body?) | LIM-9, UNC-98, CPNA-1, PXL-1, LIM-8, MYO-3, CSN-5, UNC-15 | Unc, accumulations of UNC-15, UNC-98, CSN-5 | Mercer et al. 2006 | |
| UNC-97 | PINCH | M-line, dense body (base), nucleus | PAT-4, UNC-95, LIM-8, LIM-9, UNC-98 | Unc, Pat; in Pat embryos actin and myosin are polarized, but no thick or thin filaments | Hobert et al. 1999; Norman et al. 2007 |
| UNC-98 | M-line, nucleus, (dense body?) | UNC-97, UNC-96, MYO-3, UNC-15, CSN-5 | Unc, accumulations of UNC-15, UNC-96, CSN-5 | Mercer et al. 2003; Miller et al. 2006 | |
| UNC-112 | kindlin | M-line, dense body (base) | PAT-3, PAT-4, UIG-1, DIM-1 | Unc, Pat; in Pat embryos actin and myosin not polarized | Rogalski et al. 2000; Qadota et al. 2012 |
| UNC-120 | serum response factor | nuclei? | intragenic deletion is embryonic lethal; ts allele at 20-25 shows progressive paralysis and reduced numbers of A and I bands and small fraction Pat | Baugh et al. 2005; Fukushige et al. 2006 | |
| ZYX-1 | zyxin | M-line, dense body (middle), nucleus | LIM-9, LIM-8, SCPL-1, UIG-1, ALP-1, DYC-1, DEB-1, ATN-1 | reduced maximal bending; suppresses muscle degeneration in dys-1; hlh-1 | Lecroisey et al. 2008; Lecroisey et al. 2013 |
|
*: actin regulatory proteins not included; please see Ono 2014 ?: interactions inferred from what is known about vertebrate orthologs and/or genetics |
|||||
The authors thank Claire Lecroisey, Nicolas Brouilly and Mathieu Baritaud for designing Figures 2, 5, and 11, respectively, and Nicole Mounier for helpful comments and suggestions. The authors gratefully acknowledge the very helpful suggestions for improving the manuscript from the reviewers (Don Moerman, and Michael Krause), and the editors, Michel Labouesse and Jane Mendel. K.G. acknowledges grant support from the Claude Bernard University Lyon1 and the CNRS, the Association Française contre les Myopathies and the French Agence Nationale de la Recherche. G.M.B. acknowledges grant support from the NIH (AR064307) and the Human Frontier Science Program (RGP0044/2012).
Ahringer, J. (1997). Maternal control of a zygotic patterning gene in Caenorhabditis elegans. Development 124, 3865-3869. Abstract
Albertson, D.G. (1985). Mapping muscle protein genes by in situ hybridization using biotin-labeled probes. EMBO J. 4, 2493-2498. Abstract
Ao, W., and Pilgrim, D. (2000). Caenorhabditis elegans UNC-45 is a component of muscle thick filaments and colocalizes with myosin heavy chain B, but not myosin heavy chain A. J. Cell Biol. 148, 375-384. Abstract Article
Ardizzi, J.P., and Epstein, H.F. (1987). Immunochemical localization of myosin heavy chain isoforms and paramyosin in developmentally and structurally diverse muscle cell types of the nematode Caenorhabditis elegans. J. Cell Biol. 105, 2763-2770. Abstract Article
Bagnato, P., Barone, V., Giacomello, E., Rossi, D., and Sorrentino, V. (2003). Binding of an ankyrin-1 isoform to obscurin suggests a molecular link between the sarcoplasmic reticulum and myofibrils in striated muscles. J. Cell Biol. 160, 245-253. Abstract Article
Bang, M.L., Centner, T., Fornoff, F., Geach, A.J., Gotthardt, M., McNabb, M., Witt, C.C., Labeit, D., Gregorio, C.C., Granzier, H., and Labeit, S. (2001). The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89, 1065-1072. Abstract Article
Barral, J.M., Bauer, C.C., Ortiz, I., and Epstein, H.F. (1998). unc-45 mutations in Caenorhabditis elegans implicate a CRO1/She4p-like domain in myosin assembly. J. Cell Biol. 143, 1215-1225. Abstract Article
Barral, J.M., Hutagalung, A.H., Brinker, A., Hartl, F.U., and Epstein, H.F. (2002). Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science 295, 669-671. Abstract Article
Barstead, R.J., and Waterston, R.H. (1989). The basal component of the nematode dense body is vinculin. J. Biol. Chem. 264, 10177-10185. Abstract
Barstead, R.J., Kleiman, L., and Waterston, R.H. (1991). Cloning, sequencing and mapping of an α-actinin gene from the nematode C. elegans. Cell Motil. Cytoskeleton 20, 69-78. Abstract Article
Barstead, R.J., and Waterston, R.H. (1991). Vinculin is essential for muscle function in the nematode. J. Cell Biol. 114, 715-724. Abstract Article
Baugh, L.R., Wen J.C., Hill A.A., Slonim D.K., Brown E.L., and Hunter C.P. (2005). Synthetic lethal analysis of Caenorhabditis elegans posterior embryonic patterning genes identifies conserved genetic interactions. Genome Biol. 6, R45. Abstract Article
Baugh, L.R., and Hunter, C.P. (2006). MyoD, modularity, and myogenesis: conservation of regulators and redundancy in C. elegans. Genes Dev. 20, 3342-3346. Abstract Article
Benian, G.M., Kiff, J.E., Neckelmann, N., Moerman, D.G., and Waterston, R.H. (1989). Sequence of an unusually large protein implicated in regulation of myosin activity in C. elegans. Nature 342, 45-50. Abstract Article
Benian, G.M., L'Hernault S.W., and Morris, M.E. (1993). Additional sequence complexity in the muscle gene, unc-22, and its encoded protein, twitchin, of Caenorhabditis elegans. Genetics 134, 1097-1104. Abstract
Benian, G.M., Tinley, T.L., Tang, X., and Borodovsky, M. (1996). The Caenorhabditis elegans gene unc-89, required for muscle M-line assembly, encodes a giant modular protein composed of Ig and signal transduction domains. J. Cell Biol. 132, 835-848. Abstract Article
Benian, G.M., Ayme-Southgate, A., and Tinley, T.L. (1999). The genetics and molecular biology of the titin/connectin-like proteins of invertebrates. Rev. Physiol. Biochem. Pharmacol. 138, 235-268. Abstract Article
Bessou, C., Giugia, J.B., Franks, C.J., Holden-Dye, L., and Ségalat, L. (1998). Mutations in the Caenorhabditis elegans dystrophin-like gene dys-1 lead to hyperactivity and suggest a link with cholinergic transmission. Neurogenetics 2, 61–72. Abstract Article
Blake, D.J., Weir, A., Newey, S.E., and Davies, K.E. (2002). Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol. Rev. 82, 291–329. Abstract Article
Bodine, S.C., Latres, E., Baumhueter, S., Lai, V.K., Nunez, L., Clarke, B.A., Poueymirou, W.T., Panaro, F.J., Na, E., Dharmarajan, K., et al. (2001). Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704-1708. Abstract Article
Bogomolovas, J., Gasch, A., Simkovic, F., Rigden, D.J., Labeit, S., and Mayans, O. (2014). Titin kinase is an inactive pseudokinase scaffold that supports MuRF1 recruitment to the sarcomeric M-line. Open Biol. 4, 140041. Abstract Article
Bosu, D.R., and Kipreos, E.T. (2008). Cullin-RING ubiquitin ligases: global regulation and activation cycles. Cell Div. 3, doi:10.1186/1747-1028-3-7 Abstract Article
Bowerman, B., Eaton B.A., and Priess J.R. (1992). skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell 68, 1061-1075. Abstract Article
Bowerman, B., Ingram M.K., Hunter C.P. (1997). The maternal par genes and the segregation of cell fate specification activities in early Caenorhabditis elegans embryos. Development 124, 3815-3826. Abstract
Bowman, A.L., Kontrogianni-Konstantopoulos, A., Hirsch, S.S., Geisler, S.B., Gonzalez-Serratos, H., Russell, M.W., and Bloch, R.J. (2007). Different obscurin isoforms localize to distinct sites at sarcomeres. FEBS Lett. 581, 1549-54. Abstract Article
Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. Abstract
Broday, L., Kolotuev, I., Didier, C., Bhoumik, A., Podbilewicz, B., and Ronai, Z. (2004). The LIM domain protein UNC-95 is required for the assembly of muscle attachment structures and is regulated by the RING finger protein RNF-5 in C. elegans. J. Cell Biol. 165, 857-867. Abstract Article
Brussee, V., Tardif, F., and Tremblay, J.P. (1997). Muscle fibers of mdx mice are more vulnerable to exercise than those of normal mice. Neuromuscul. Disord. 7, 487–492. Abstract Article
Brouilly, N., Lecroisey, C., Martin, E., Pierson, L., Mariol, M.-C., Qadota, H., Labouesse, M., Streichenberger, N., Mounier, N., and Gieseler, K. (2015). Ultra-structural time-course study in the C. elegans model for Duchenne muscular dystrophy highlights a crucial role for sarcomere-anchoring structures and sarcolemma integrity in the earliest steps of the muscle degeneration process. Hum. Mol. Genet. 24, 6428–6445. Abstract Article
Brown, A.E., Yemini, E.I., Grundy, L.J., Jucikas, T., and Schafer, W.R. (2013). A dictionary of behavioral motifs reveals clusters of genes affecting Caenorhabditis elegans locomotion. Proc. Natl. Acad. Sci. U. S. A. 110, 791-796. Abstract Article
Burkeen, A.K. Maday, S.L., Rybicka, K.K., Sulcove, J.A., Ward, J., Huang, M.M., Barstead, R., Franzini-Armstrong, C., and Allen, T.S. (2004). Disruption of Caenorhabditis elegans muscle structure and function caused by mutation of troponin I. Biophys. J. 86, 991–1001. Abstract Article
Butler, T.M. and Siegman, M.J. (2011). A force-activated kinase in a catch smooth muscle. J. Muscle Res. Cell Motil. 31, 349-358. Abstract Article
Carmeli, E., Aizenbud, D., and Rom, O. (2015). How do skeletal muscles die? An overview. Adv. Exp. Med. Biol. 14, 99–111. Abstract Article
Carre-Pierrat, M., Grisoni, K., Gieseler, K., Mariol, M.-C., Martin, E., Jospin, M., Allard, B., and Ségalat, L. (2006). The SLO-1 BK channel of Caenorhabditis elegans is critical for muscle function and is involved in dystrophin-dependent muscle dystrophy. J. Mol. Biol. 358, 387–395. Abstract Article
Chen, L., Krause M., Draper B., Weintraub H., and Fire A. (1992). Body-wall muscle formation in Caenorhabditis elegans embryos that lack the MyoD homolog hlh-1. Science 256, 240-243. Abstract Article
Chen, L., Krause M., Sepanski M., and Fire A. (1994). The Caenorhabditis elegans MYOD homologue HLH-1 is essential for proper muscle function and complete morphogenesis. Development 120, 1631-1641. Abstract
Chow, D., Srikakulam, R., Chen, Y., and Winkelmann, D.A. (2002). Folding of the striated muscle myosin motor domain. J. Biol. Chem. 277, 36799-36807. Abstract Article
Constantin, B. (2014). Dystrophin complex functions as a scaffold for signalling proteins. Biochim. Biophys. Acta 1838, 635–642. Abstract Article
Cope, G.A., and Deshaies, R.J. (2003). COP9 signalosome: multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell 114, 663-671. Abstract Article
Corsi, A.K., Kostas S.A., Fire A., and Krause M. (2000). Caenorhabditis elegans twist plays an essential role in non-striated muscle development. Development 127, 2041-2051. Abstract
Cox, E.A., and Hardin, J. (2004). Sticky worms: adhesion complexes in C. elegans. J. Cell Sci. 117, 1885-1897. Abstract Article
Deitiker, P.R., and Epstein, H.F. (1993). Thick filament substructures in C. elegans: evidence for two populations of paramyosin. J. Cell Biol. 123, 303–311. Abstract Article
De, Luca, A., Pierno, S., Liantonio, A., Cetrone, M., Camerino, C., Fraysse, B., Mirabella, M., Servidei, S., Rüegg, U.T., and Conte Camerino, D. (2003). Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J. Pharmacol. Exp. Ther. 304, 453–463. Abstract Article
Dey, C.S., Deitiker, P.R., and Epstein, H.F. (1992). Assembly-dependent phosphorylation of myosin and paramyosin of native thick filaments in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 186, 1528-1532. Abstract Article
Dibb, N.J., Brown, D.M., Karn, J., Moerman, D.G., Bolten, S.L., and Waterston, R.H. (1985). Sequence analysis of mutations that affect the synthesis, assembly and enzymatic activity of the unc-54 myosin heavy chain of Caenorhabditis elegans. J. Mol. Biol. 183, 543-551. Abstract Article
Dichoso, D., Brodigan T., Chwoe K.Y., Lee J.S., Llacer R., Park M., Corsi A.K., Kostas S.A., Fire A., Ahnn J., and Krause M. (2000). The MADS-Box factor CeMEF2 is not essential for Caenorhabditis elegans myogenesis and development. Dev. Biol. 223, 431-440. Abstract Article
Didier, C., Broday, L., Bhoumik, A., Israeli, S., Takahashi, S., Nakayama, K., Thomas, S.M., Turner, C.E., Henderson, S., Sabe, H., and Ronai, Z. (2003). RNF5, a RING finger protein that regulates cell motility by targeting paxillin ubiquitination and altered localization. Mol. Cell. Biol. 23, 5331-5345. Abstract Article
Ding, L., and Candido, E.P. (2000). HSP25, a small heat shock protein associated with dense bodies and M-lines of body wall muscle in Caenorhabditis elegans. J. Biol. Chem. 275, 9510-9517. Abstract Article
Dixon, S.J., and Roy, P.J. (2005). Muscle arm development in Caenorhabditis elegans. Development 132, 3079–3092. Abstract Article
Edgar, L.G., Carr S., Wang H., and Wood W.B. (2001). Zygotic expression of the caudal homolog pal-1 is required for posterior patterning in Caenorhabditis elegans embryogenesis. Dev. Biol. 229, 71-88. Abstract Article
Emery, A.E. (1991). Population frequencies of inherited neuromuscular diseases–a world survey. Neuromuscul. Disord. 1, 19–29. Abstract Article
Epstein, H.F., and Thomson, J.N. (1974). Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature 250, 579-580. Abstract Article
Epstein, H.F., Waterston, R.H., and Brenner, S. (1974). A mutant affecting the heavy chain of myosin in Caenorhabditis elegans. J. Mol. Biol. 90, 291-300. Abstract Article
Epstein, H.F., Casey, D.L., and Ortiz, I. (1993). Myosin and paramyosin of Caenorhabditis elegans embryos assemble into nascent structures distinct from thick filaments and multi-filament assemblages. J. Cell Biol. 122, 845–858. Abstract Article
Epstein, H.F., Lu, G.Y., Deitiker, P.R., Ortiz, I., and Schmid, M.F. (1995). Preliminary three-dimensional model for nematode thick filament core. J. Struct. Biol. 115, 163-174. Abstract Article
Epstein, H.F., and Benian, G.M. (2012). Paradigm shifts in cardiovascular research from Caenorhabditis elegans muscle. Trends Cardiovasc. Med. 22, 201-209. Abstract Article
Ervasti, J.M. (2003). Costameres: the Achilles’ heel of Herculean muscle. J. Biol. Chem. 278, 13591-13594. Abstract Article
Etheridge, T., Oczypok, E.A., Lehmann, S., Fields, B.D., Shephard, F., Jacobson, L.A., and Scewczyk, N.J. (2012). Calpains mediate integrin attachment complex maintenance of adult muscle in Caenorhabditis elegans. PLoS Genet. 8, e1002471. Abstract Article
Etheridge, T., Rahman, M., Gaffney, C.J., Shaw, D., Shephard, F., Magudia, J., Solomon, D.E., Milne, T., Blawzdziewicz, J., Constantin-Teodosiu, D., et al. (2015). The integrin-adhesome is required to maintain muscle structure, mitochondrial ATP production, and movement forces in Caenorhabditis elegans. FASEB J. 29, 1235-1246. Abstract Article
Ferrara, T.M., Flaherty, D.B., and Benian, G.M. (2005). Titin/connectin-related proteins in C. elegans: a review and new findings. J. Muscle Res. Cell Motil. 26, 435-447. Abstract Article
Flaherty, D.B., Gernert, K.M., Shmeleva, N., Tang, X., Mercer, K.B., Borodovsky, M., and Benian, G.M. (2002). Titins in C. elegans with unusual features: coiled-coil domains, novel regulation of kinase activity and two new possible elastic regions. J. Mol. Biol. 323, 533-549. Abstract Article
Forbes, J.G., Flaherty, D.B., Ma, K., Qadota, H., Benian, G.M., and Wang, K. (2010). Extensive and modular intrinsically disordered segments in C. elegans TTN-1 and implications in filament binding, elasticity and oblique striation. J. Mol. Biol. 398, 672-689. Abstract
Ford-Speelman, D.L., Roche, J.A., Bowman, A.L., and Bloch, R.J. (2009). The rho-guanine nucleotide exchange domain of obscurin activates rhoA signaling in skeletal muscle. Mol. Biol. Cell 20, 3905-3917. Abstract
Francis, G.R., and Waterston, R.H. (1985). Muscle organization in Caenorhabditis elegans: localization of proteins implicated in thin filament attachment and I-band organization. J. Cell Biol. 101, 1532-1549. Abstract Article
Francis, R., and Waterston, R.H. (1991). Muscle cell attachment in Caenorhabditis elegans. J. Cell Biol. 114, 465-479. Abstract Article
Fukuda, K., Gupta, S., Chen, K., Wu, C., and Qin, J. (2009). The pseudoactive site of ILK is essential for its binding to α-Parvin and localization to focal adhesions. Mol. Cell 36, 819-830. Abstract Article
Fukushige, T. and Krause M. (2005). The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development 132, 1795-1805. Abstract Article
Fukushige, T., Brodigan T.M., Schriefer L.A., Waterston R.H., and Krause M. (2006). Defining the transcriptional redundancy of early bodywall muscle development in C. elegans: evidence for a unified theory of animal muscle development. Genes Dev. 20, 3395-3406. Abstract Article
Funabara, D., Hamamoto, C., Yamamoto, K., Inoue, A., Ueda, M., Osawa, R., Kanoh, S., Hartshorne, D.J., Suzuki, S., and Watabe, S. (2007). Unphosphorylated twitchin forms a complex with actin and myosin that may contribute to tension maintenance in catch. J. Exp. Biol. 210, 4399-4410. Abstract Article
Furukawa, M., He, Y.J., Borchers, C., and Xiong, Y. (2003). Targeting of protein ubiquitination by BTB-cullin 3-Roc1 ubiquitin ligases. Nat. Cell Biol. 5, 1001-1007. Abstract
Gaiser, A.M., Kaiser, C.J.O., Haslbeck, V., and Richter, K. (2011). Downregulation of the Hsp90 system causes defects in muscle cells of Caenorhabditis elegans. PLoS One 6, e25485. Abstract
Gautel, M. (2008). The sarcomere and the nucleus: functional links to hypertrophy, atrophy and sarcopenia. Adv. Exp. Med. Biol. 642, 176-191. Abstract Article
Gazda, L., Pokrzywa, W., Hellerschmied, D., Lowe, T., Forne, I., Mueller-Planitz, F., Hoppe, T., and Clausen, T. (2013). The myosin chaperone UNC-45 is organized in tandem modules to support myofilament formation in C. elegans. Cell 152, 183-195. Abstract Article
Gettner, S.N., Kenyon, C., and Reichardt, L.F. (1995). Characterization of βpat-3 heterodimers, a family of essential integrin receptors in C. elegans. J. Cell Biol. 129, 1127-1141. Abstract Article
Ghosh, S.R., and Hope, I.A. (2010). Determination of the mobility of novel and established Caenorhabditis elegans sarcomeric proteins in vivo. Eur. J. Cell Biol. 89, 437-448. Abstract Article
Giacomotto, J., Brouilly, N., Walter, L., Mariol, M.-C., Berger, J., Ségalat, L., Becker, T.S., Currie, P.D., and Gieseler, K. (2013). Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Hum. Mol. Genet. 22, 4562-4578. Abstract
Gieseler, K., Bessou, C., and Ségalat, L. (1999a). Dystrobrevin- and dystrophin-like mutants display similar phenotypes in the nematode Caenorhabditis elegans. Neurogenetics 2, 87–90. Abstract Article
Gieseler, K., Abdel-Dayem, M., and Ségalat, L. (1999b). In vitro interactions of Caenorhabditis elegans dystrophin with dystrobrevin and syntrophin. FEBS Lett. 461, 59–62. Abstract Article
Gieseler, K., Grisoni, K., and Ségalat, L. (2000). Genetic suppression of phenotypes arising from mutations in dystrophin-related genes in Caenorhabditis elegans. Curr. Biol. 10, 1092–1097. Abstract Article
Giugia, J., Gieseler, K., Arpagaus, M., and Ségalat, L. (1999). Mutations in the dystrophin-like dys-1 gene of Caenorhabditis elegans result in reduced acetylcholinesterase activity. FEBS Lett. 463, 270–272. Abstract Article
Gomes, M.D., Lecker, S.H., Jagoe, R.T., Navon, A., and Goldberg, A.L. (2001). Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. U. S. A. 98, 14440-14445. Abstract Article
Grater, F., Shen, J., Jiang, H., Gautel, M., and Grubmuller, H. (2005). Mechanically induced titin kinase activation studied by force-probe molecular dynamics simulations. Biophys. J. 88, 790-804. Abstract Article
Greene, D.N., Garcia, T., Sutton, R.B., Gernert, K.M., Benian, G.M., and Oberhauser, A.F. (2008). Single-molecule force spectroscopy reveals a stepwise unfolding of Caenorhabditis elegans giant protein kinase domains. Biophys. J. 95, 1360-1370. Abstract
Grisoni, K., Martin, E., Gieseler, K., Mariol, M.-C., and Ségalat, L. (2002). Genetic evidence for a dystrophin-glycoprotein complex (DGC) in Caenorhabditis elegans. Gene 294, 77–86. Abstract
Grisoni, K., Gieseler, K., Mariol, M.C., Martin, E., Carre-Pierrat, M., Moulder, G., Barstead, R., and Ségalat, L. (2003). The stn-1 syntrophin gene of C. elegans is functionally related to dystrophin and dystrobrevin. J. Mol. Biol. 332, 1037–1046. Abstract Article
Han, H.F., and Beckerle, M.C. (2009). The ALP-Enigma protein ALP-1 functions in actin filament organization to promote muscle structural integrity in Caenorhabditis elegans. Mol. Biol. Cell 20, 2361-2370. Abstract Article
Harfe, B.D., Branda, C.S., Krause, M., Stern, M.J., and Fire, A. (1998). MyoD and the specification of muscle and non-muscle fates during postembryonic development of the C. elegans mesoderm. Development 125, 2479-2488. Abstract
Harfe, B.D., Vaz Gomes A., Kenyon C., Liu J., Krause M., and Fire A. (1998). Analysis of a Caenorhabditis elegans Twist homolog identifies conserved and divergent aspects of mesodermal patterning. Genes Dev. 12, 2623-2635. Abstract Article
Herndon, L. A., Schmeissner, P. J., Dudaronek, J. M., Brown, P. A., Listner, K. M., Sakano, Y., Paupard, M.C., Hall, D.H., and Driscoll, M. (2002). Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419, 808–814. Abstract Article
Hikita, T., Qadota, H., Tsuboi, D., Taya, S., Moerman, D.G., and Kaibuchi, K. (2005). Identification of a novel Cdc42 GEF that is localized to the PAT-3-mediated adhesive structure. Biochem. Biophys. Res. Commun. 335, 139-145. Abstract Article
Hobert, O., Moerman, D.G., Clark, K.A., Beckerle, M.C., and Ruvkun, G. (1999). A conserved LIM protein that affects muscular adherens junction integrity and mechanosensory function in Caenorhabditis elegans. J. Cell Biol. 144, 45-57. Abstract Article
Hoffman, E.P., Brown, R.H., Jr, and Kunkel, L.M. (1987). Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51, 919–928. Abstract Article
Hoppe, P.E., Chau, J., Flanagan, K.A., Reedy, A.R., and Schriefer, L.A. (2010). Caenorhabditis elegans unc-82 encodes a serine-threonine kinase important for myosin filament organization in muscle during growth. Genetics 184, 79-90. Abstract Article
Hresko, M.C., Williams, B.D., and Waterston, R.H. (1994). Assembly of body wall muscle and muscle cell attachment structures in Caenorhabditis elegans. J. Cell Biol. 124, 491-506. Abstract Article
Hu, S.H., Parker, M.W., Lei, J.Y., Wilce, M.C., Benian, G.M., and Kemp, B.E. (1994). Insights into autoregulation from the crystal structure of twitchin kinase. Nature 369, 581-584. Abstract Article
Hu, L.Y., and Kontrogianni-Konstantopoulos, A. (2013). The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J. 27, 2001-2012. Abstract Article
Hunter, C.P. and Kenyon C. (1996). Spatial and temporal controls target pal-1 blastomere-specification activity to a single blastomere lineage in C. elegans embryos. Cell. 87, 217-226. Abstract Article
Husson, S.J. et al. Keeping track of worm trackers (September 10, 2012), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.156.1, http://www.wormbook.org. Abstract
Hwang, H., Barnes, D.E., Matsunaga, Y., Benian, G.M., Ono, S., and Lu, H. (2016). Muscle contraction phenotypic analysis enabled by optogenetics reveals functional relationships of sarcomere components in Caenorhabditis elegans. Sci. Rep. 6, 19900. Abstract Article
Johari, S., Nock, V., Alkaisi, M.M., and Wang, W. (2013). On-chip analysis of C. elegans muscular forces and locomotion patterns in microstructured environments. Lab Chip 13, 1699-1707. Abstract Article
Johnson, R.P., Kang, S.H., and Kramer, J.M. (2006). C. elegans dystroglycan DGN-1 functions in epithelia and neurons, but not muscle, and independently of dystrophin. Development 133, 1911–1921. Abstract Article
Kachur, T., Ao, W., Berger, J., and Pilgrim, D. (2004). Maternal UNC-45 is involved in cytokinesis and colocalizes with non-muscle myosin in the early Caenorhabditis elegans embryo. J. Cell Sci. 117, 5313-5321. Abstract Article
Kachur, T.M., Audhya, A., and Pilgrim, D.B. (2008). UNC-45 is required for NMY-2 contractile function in early embryonic polarity establishment and germline cellularization in C. elegans. Dev. Biol. 314, 287-299. Abstract Article
Kagawa, H., Gengyo, K., McLachlan, A.D., Brenner, S., and Karn, J. (1989). Paramyosin gene (unc-15) of Caenorhabditis elegans. Molecular cloning, nucleotide sequence and models for thick filament structure. J. Mol. Biol. 207, 311-333. Abstract Article
Kaiser, C.M., Bujalowski, P.J., Ma, L., Anderson, J., Epstein, H.F., and Oberhauser, A.F. (2012). Tracking UNC-45 chaperone-myosin interaction with a titin mechanical reporter. Biophys. J. 102, 2212- 2219. Abstract Article
Karn, J., Brenner, S., and Barnett, L. (1983). Protein structural domains in the Caenorhabditis elegans unc-54 myosin heavy chain gene are not separated by introns. Proc. Natl. Acad. Sci. U. S. A. 80, 4253-4257. Abstract Article
Kim, H., Rogers, M.J., Richmond, J.E., and McIntire, S.L. (2004). SNF-6 is an acetylcholine transporter interacting with the dystrophin complex in Caenorhabditis elegans. Nature 430, 891–896. Abstract Article
Kim, H., Pierce-Shimomura, J.T., Oh, H.J., Johnson, B.E., Goodman, M.B., and McIntire, S.L. (2009). The dystrophin complex controls BK channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet. 5, e1000780. Abstract Article
Knappeis, G.G., and Carlsen, F. (1968). The ultrastructure of the M line in skeletal muscle. J. Cell Biol. 38, 202-211. Abstract Article
Kobe, B., Heierhorst, J., Feil, S.C., Parker, M.W., Benian, G.M., Weiss, K.R., and Kemp, B.E. (1996). Giant protein kinases: domain interactions and structural basis of autoregulation. EMBO J. 15, 6810-6821. Abstract
Koenig, M., Hoffman, E.P., Bertelson, C.J., Monaco, A.P., Feener, C., and Kunkel, L.M. (1987). Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50, 509–517. Abstract Article
Kontrogianni-Konstantopoulos, A., Jones, E.M., Van Rossum, D.B., and Bloch, R.J. (2003). Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138-1148. Abstract Article
Koren, Y., Sznitman, R., Arratia, P.E., Caris, C., Krajacic, P., Brown, A.E.X., and Sznitman, J. (2015). Model-independent phenotyping of C. elegans locomotion using scale-invariant feature transform. PLoS One 10, e0122326. Abstract Article
Krajacic, P., Shen, X., Purohit, P.K., Arratia, P., and Lamitina, T. (2012). Biomechanical profiling of Caenorhabditis elegans motility. Genetics 191, 1015-1021. Abstract Article
Krause, M., Fire A., Harrison S.W., Priess J, and Weintraub H. (1990). CeMyoD accumulation defines the body wall muscle cell fate during C. elegans embryogenesis. Cell 63, 907-919. Abstract Article
Krause, M., Harrison S.W., Xu S.Q., Chen L., and Fire A. (1994). Elements regulating cell- and stage-specific expression of the C. elegans MyoD family homolog hlh-1. Dev. Biol. 166, 133-148. Abstract Article
Kuo, W.-J., Sie, Y.-S., and Chuang, H.-S. (2014). Characterizations of kinetic power and propulsion of the nematode Caenorhabditis elegans based on a micro-particle image velocimetry system. Biomicrofluidics 8, 024116. Abstract Article
Landsverk, M.L., Li, S., Hutagalung, A.H., Najafov, A., Hoppe, T., Barral, J.M., and Epstein, H.F. (2007). The UNC-45 chaperone mediates sarcomere assembly through myosin degradation in Caenorhabditis elegans. J. Cell Biol. 177, 205-210. Abstract Article
Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E., Kristensen, J., Brandmeier, B., Franzen, G., Hedberg, B, et al. (2005). The kinase domain of titin controls muscle gene expression and protein turnover. Science 308, 1599-1603. Abstract Article
Lange, S., Ouyang, K., Meyer, G., Cui, L., Cheng, H., Lieber, R.L., and Chen, J. (2009). Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. 122, 2640-2650. Abstract Article
Lange, S., Perera, S., Teh, P., and Chen, J. (2012). Obscurin and KCTD6 regulate cullin-depenent small ankyrin-1 (sAnk1.5) protein turnover. Mol. Biol. Cell 23, 2490-2504. Abstract Article
Lebart, M.C., and Benyamin, Y. (2006). Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 273, 3415-3426. Abstract Article
Lecroisey, C. (2010). Caractérisation moléculaire et cellulaire de la dégénérescence musculaire dépendante de la dystrophine chez le nématode Caenorhabditis elegans. Thesis manuscript, University Lyon 1. Abstract
Lecroisey, C., Martin, E., Mariol, M.-C., Granger, L., Schwab, Y., Labouesse, M., Ségalat, L., and Gieseler, K. (2008). DYC-1, a protein functionally linked to dystrophin in Caenorhabditis elegans is associated with the dense body, where it interacts with the muscle LIM domain protein ZYX-1. Mol. Biol. Cell 19, 785–796. Abstract Article
Lecroisey, C., Brouilly, N., Qadota, H., Mariol, M.-C., Rochette, N.C., Martin, E., Benian, G.M., Ségalat, L., Mounier, N., and Gieseler, K. (2013). ZYX-1, the unique zyxin protein of Caenorhabditis elegans, is involved in dystrophin-dependent muscle degeneration. Mol. Biol. Cell 24, 1232–1249. Abstract Article
Lee, C.F., Hauenstein, A.V., Fleming, J.K., Gasper, W.C., Engelke, V., Sankaran, B., Bernstein, S.I., and Huxford, T. (2011). X-ray crystal structure of the UCS domain-containing UNC-45 myosin chaperone from Drosophila melanogaster. Structure 19, 397-408. Abstract Article
Lei, J., Tang, X., Chambers, T.C., Pohl, J., and Benian, G.M. (1994). Protein kinase domain of twitchin has protein kinase activity and an autoinhibitory region. J. Biol. Chem. 269, 21078-21085. Abstract
Lei, H., Fukushige T., Niu W., Sarov M., Reinke V., and Krause M. (2010). A widespread distribution of genomic CeMyoD binding sites revealed and cross validated by ChIP-Chip and ChIP-Seq techniques. PLoS One 5, e15898. Abstract
Lei, H., Liu J., Fukushige T., Fire A,, and Krause M. (2009). Caudal-like PAL-1 directly activates the bodywall muscle module regulator hlh-1 in C. elegans to initiate the embryonic muscle gene regulatory network. Development 136, 1241-1249. Abstract Article
Lin, X., Qadota, H., Moerman, D.G. and Williams, B.D. (2003). C. elegans PAT-6/Actopaxin plays a critical role in the assembly of integrin adhesion complexes in vivo. Curr. Biol. 13, 922-932. Abstract Article
Liu, F., Bauer CC, Ortiz I, Cook RG, Schmid MF, and Epstein HF (1998). β-filagenin, a newly identified protein coassembling with myosin and paramyosin in Caenorhabditis elegans. J. Cell Biol. 140, 347-353. Abstract Article
Liu, F., Ortiz, I, Hutagalung, A., Bauer, C.C., Cook, R.G., and Epstein, H.F. (2000). Differential assembly of α- and γ-filagenins into thick filaments in Caenorhabditis elegans. J. Cell Sci. 113, 4001-4012. Abstract
Liu, J, Schrank B, and Waterston RH (1996). Interaction between a putative mechanosensory membrane channel and a collagen. Science 273, 361-364. Abstract Article
Liu, Q., Jones, T.I., Bachmann, R.A., Meghpara, M., Rogowshi, L., Williams, B.D., and Jones, P.L. (2012). C. elegans PAT-9 is a nuclear zinc finger protein critical for the assembly of muscle attachments. Cell Biosci. 2, 18. Abstract Article
Liu, Q., Jones, T.I., Tang, V.W., Brieher, W.M., and Jones, P.L. (2010). Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J. Cell Sci. 123, 1116-1123. Abstract Article
Luther, P., and Squire, J. (1978). Three-dimensional structure of the vertebrate muscle M-region. J. Mol. Biol. 125, 313-324. Abstract Article
Mackenzie, J.M., and Epstein, H.F. (1980). Paramyosin is necessary for determination of nematode thick filament length in vivo. Cell 22, 747-755. Abstract Article
Mackinnon, A.C., Qadota, H., Norman, K.R., Moerman, D.G., and Williams, B.D. (2002). C. elegans PAT-4 / ILK functions as an adaptor protein within integrin adhesion complexes. Curr. Biol. 12, 787-797. Abstract Article
Mariol, MC., Martin, E., Chambonnier L. and Ségalat L. (2007). Dystrophin-dependent muscle degeneration requires a fully functional contractile machinery to occur in C. elegans. Neuromuscul. Disord. 17, 56-60. Abstract
Mariol, MC., and Ségalat, L. (2001). Muscular degeneration in the absence of dystrophin is a calcium-dependent process. Curr Biol. 11, 1691-1694. Abstract
Maruyama, I.N., Miller, D.M., and Brenner S. (1989). Myosin heavy chain gene amplification as a suppressor mutation in Ceonorhabditis elegans. Mol. Gen. Genet. 219, 113-118. Abstract Article
Mathies, L.D., Henderson, S.T., and Kimble, J. (2003). The C. elegans Hand gene controls embryogenesis and early gonadogenesis. Development 130, 2881-2892. Abstract Article
Matsunaga, Y., Qadota, H., Furukawa, M., Choe, H.H., and Benian, G.M. (2015). Twitchin kinase interacts with MAPKAP kinase 2 in Caenorhabditis elegans striated muscle. Mol. Biol. Cell 26, 2096-2111. Abstract Article
Mayans, O., van der Ven, P.F., Wilm, M., Mues, A., Young, P., Furst, D.O., Wilmanns, M., and Gautel M. (1998). Structural basis for activation of the titin kinase domain during myofibrillogenesis. Nature 395, 863-869. Abstract Article
McDonald, K.A., Lakonishok, M., and Horwitz, A.F. (1995). αv and α3 integrin subunits are associated with myofibrils during myofibrillogenesis. J. Cell Sci. 108, 2573-2581. Abstract
McKeown, C.R., Han, H.F., and Beckerle, M.C. (2006). Molecular characterization of the Caenorhabditis elegans ALP/Enigma gene alp-1. Dev. Dyn. 235, 530-538. Abstract Article
McKim, K.S., Matheson, C., Marra, M.A., Wakarchuk, M.F., and Baillie, D.L. (1994). The Caenorhabditis elegans unc-60 gene encodes proteins homologous to a family of actin-binding proteins. Mol. Gen. Genet. 242, 346-357. Abstract Article
McLachlan, A.D., and Karn, J. (1982). Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature 299, 226-231. Abstract Article
Megeney, L., Kablar B., Garrett K., Anderson J,. Rudnicki M. (1996)..MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev, 10, 1173-1183. Abstract Article
Meissner, B., Warner, A., Wong, K., Dube, N., Lorch, A., McKay, S.J., Khattra, J., Rogalski, T., Somasiri, A., Chaudhry, I., et al. (2009). An integrated strategy to study muscle development and myofilament structure in C. elegans. PLoS Genet. 5, e1000537. Abstract Article
Meissner, B., Rogalski, T., Viveiros, R., Warner, A., Plastino, L., Lorch, A., Granger, L., Ségalat, L., and Moerman, D.G. (2011). Determining the sub-cellular localization of proteins within Caenorhabditis elegans body wall muscle. PLoS One 6, e19937. Abstract Article
Mercer, K.B., Flaherty, D.B., Miller, R.K., Qadota, H., Tinley, T.L., Moerman, D.G., and Benian, G.M. (2003). C. elegans UNC-98, a C2H2 Zn finger protein, is a novel partner of UNC-97/PINCH in muscle adhesion complexes. Mol. Biol. Cell 14, 2492-2507. Abstract Article
Mercer, K.B., Miller, R.K., Tinley, T.L., Sheth, S., Qadota, H., and Benian, G.M. (2006). Caenorhabditis elegans UNC-96 is a new component of M-lines that interacts with UNC-98 and paramyosin and is required in adult muscle for assembly and/or maintenance of thick filaments. Mol. Biol. Cell 17, 3832-3847. Abstract Article
Miller, D.M. 3rd, Ortiz, I., Berliner, G.C., and Epstein, H.F. (1983). Differential localization of two myosins within nematode thick filaments. Cell 34, 477-490. Abstract Article
Miller, D.M. 3rd, Stockdale, F.E., and Karn, J. (1986). Immunological identification of the genes encoding the four myosin heavy chain isoforms of Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 83, 2305-2309. Abstract Article
Miller, R.K., Qadota, H., Landsverk, M.L., Mercer, K.B., Epstein, H.F., and Benian, G.M. (2006). UNC-98 links an integrin-associated complex to thick filaments in Caenorhabditis elegans muscle. J. Cell Biol. 175, 853-859. Abstract Article
Miller, R.K., Qadota, H., Mercer, K.B., Gernert, K.M., and Benian, G.M. (2008). UNC-98 and UNC-96 interact with paramyosin to promote its incorporation into thick filaments of Caenorhabditis elegans. Mol. Biol. Cell 19, 1529-1539. Abstract Article
Miller, R.K., Qadota, H., Stark, T.J., Mercer, K.B., Wortham, T.S., Anyanful, A., and Benian, G.M. (2009). CSN-5, a component of the COP9 signalosome complex, regulates the levels of UNC-96 and UNC-98, two components of M-lines in Caenorhabditis elegans muscle. Mol. Biol. Cell 20, 3608-3616. Abstract Article
Mi-Mi, L., Votra, S.B., Kemphues, K., Bretscher, A., and Pruyne, D. (2012). Z-line formins promote contractile lattice growth and maintenance in striated muscles of C. elegans. J. Cell Biol. 198, 87-102. Abstract Article
Moerman, D.G., Plurad, S., Waterston, R.H., and Baillie, D.L. (1982). Mutations in the unc-54 myosin heavy chain gene of Caenorhabditis elegans that alter contractility but not muscle structure. Cell 29, 773–781. Abstract Article
Moerman, D.G., Benian, G.M., and Waterston, R.H. (1986). Molecular cloning of the muscle gene unc-22 in Caenorhabditis elegans by Tc1 transposon tagging. Proc. Natl. Acad. Sci. U. S. A. 83, 2579-2583. Abstract Article
Moerman, D.G., Benian, G.M., Barstead, R.J., Schriefer, L.A., and Waterston, R.H. (1988). Identification and intracellular localization of the unc-22 gene product of Caenorhabditis elegans. Genes Dev. 2, 93-105. Abstract Article
Moerman, D.G., and Fire, A. (1997). Muscle Structure, Function and Development. In C. elegans II, D. L. Riddle, T. Blumenthal, B. J. Meyer, and J. R. Priess, eds. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press), pp. 417-470. Abstract
Moerman, D.G. and Williams, B.D. Sarcomere assembly in C. elegans muscle (January 16, 2006), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.81.1, http://www.wormbook.org.
Mokhtarian, A., Lefaucheur, J.P., Even, P.C., and Sebille, A. (1999). Hindlimb immobilization applied to 21-day-old mdx mice prevents the occurrence of muscle degeneration. J. Appl. Physiol. 86, 924–931. Abstract
Morley, J.F., Brignull, H.R., Weyers, J.J., and Morimoto, R.I. (2002). The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 99, 10417–10422. Abstract Article
Moulder, G.L., Huang, M.M., Waterston, R.H., and Barstead, R.J. (1996). Talin requires β-integrin, but not vinculin, for its assembly into focal adhesion-like structures in the nematode C. elegans. Mol. Biol. Cell 7, 1181-1193. Abstract Article
Moulder, G.L., Cremona, G.H., Duerr, J., Stirman, J.N., Fields, S.D., Martin, W., Qadota, H., Benian, G.M., Lu, H., and Barstead, R.J. (2010). α-actinin is required for proper assembly of Z-disk/focal adhesion-like structures and for efficient locomotion in C. elegans. J. Mol. Biol. 403, 516-528. Abstract Article
Mullen, G.P., Rogalski, T.M., Bush, J.A., Gorji, P.R., and Moerman, D.G. (1999). Complex patterns of alternatie splicing mediate the spatial and temporal distribution of perlecan/UNC-52 in Caenorhabditis elegans. Mol. Biol. Cell 10, 3205-3221. Abstract Article
Muller, S.A., Haner, M., Ortiz, I., Aebi, U., and Epstein, H.F. (2001). STEM analysis of Caenorhabditis elegans muscle thick filaments: evidence for microdifferentiated substructures. J. Mol. Biol. 305, 1035-1044. Abstract Article
Nahabedian, J.F., Qadota, H., Stirman, J.N., Lu, H., and Benian, G.M. (2012). Bending amplitude—a new quantitative assay of C. elegans locomotion: identification of phenotypes for mutants in genes encoding muscle focal adhesion components. Methods 56, 95-102. Abstract Article
Ni, W., Hutagalung, A.H., Li, S., and Epstein, H.F. (2011). The myosin-binding UCS domain but not the Hsp90-binding TPR domain of the UNC-45 chaperone is essential for function in Caenorhabditis elegans. J. Cell Sci. 124, 3164-3173. Abstract Article
Nicholls, P., Bujalowski, P.J., Epstein, H.F., Boehning, D.F., Barral, J.M., and Oberhauser, A.F. (2014). Chaperone-mediated reversible inhibition of the sarcomeric myosin power stroke. FEBS Lett. 588, 3977- 3981. Abstract Article
Norman, K.R., Fazzio, R.T., Mellem, J.E., Espelt, M.V., Strange, K., Beckerle, M.C., and Maricq, A.V. (2005). The Rho/Rac-family guanine nucleotide exchange factor VAV-1 regulates rhythmic behaviors in C. elegans. Cell 123, 119-132. Abstract Article
Norman, K.R., Cordes, S., Qadota, H., Rahmani, P., and Moerman, D.G. (2007). UNC-97/PINCH is involved in the assembly of integrin cell adhesion complexes in Caenorhabditis elegans body wall muscle. Dev. Biol. 309, 45-55. Abstract Article
Oh, K.H., and Kim, H. (2013). Reduced IGF signaling prevents muscle cell death in a Caenorhabditis elegans model of muscular dystrophy. Proc. Natl. Acad. Sci. U. S. A. 110, 19024–19029. Abstract Article
Ono, S., Baillie D.L., and Benian, G.M. (1999). UNC-60B, an ADF/cofilin family protein, is required for proper assembly of actin into myofibrils in Caenorhabditis elegans body wall muscle. J. Cell Biol. 145, 591-502. Abstract Article
Ono, K., Yu, R., Mohri, K., and Ono, S. (2006). Caenorhabditis elegans kettin, a large immunoglobulin-like repeat protein, binds to filamentous actin and provides mechanical stability to the contractile apparatuses in body wall muscle. Mol. Biol. Cell 17, 2722-2734. Abstract Article
Ono, S. (2014). Regulation of structure and function of sarcomeric actin filaments in striated muscle of the nematode Caenorhabditis elegans. Anat. Rec. 297, 1548-1559. Abstract Article
Papsdorf, K., Sacherl, J., and Richter, K. (2014). The balanced regulation of Hsc70 by DNJ-13 and UNC-23 is required for muscle functionality. J. Biol. Chem. 289, 25250-25261. Abstract Article
Park, E.C., and Horvitz, H.R. (1986). C. elegans unc-105 mutations affect muscle and are suppressed by other mutations that affect muscle. Genetics 113, 853-867. Abstract
Perry, S.V. (1999). Troponin I: inhibitor or facilitator. Mol. Cell. Biochem. 190, 9–32. Abstract Article
Pilgram, G.S., Potikanond, S., Baines, R.A., Fradkin, L.G., and Noordermeer, J.N. (2010). The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol. Neurobiol. 41, 1–21. Abstract Article
Pintard, L., Willis, J.H., Willems, A., Johnson, J.-L.F., Srayko, M., Kurz, T., Glaser, S., Mains, P.E., Tyers, M., Bowerman, B., and Peter, M. (2003). The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin ligase. Nature 425, 311-316. Abstract Article
Plenefisch, J.D., Zhu, X., and Hedgecock, E.M. (2000). Fragile skeletal muscle attachments in dystrophic mutants of Caenorhabditis elegans: isolation and characterization of the mua genes. Development 127, 1197-1207. Abstract
Polet, D., Lambrechts, A., Ono, K., Mah, A., Peelman, F., Vandekerckhove, J., Baillie, D.L., Ampe, C., and Ono, S. (2006). Caenorhabditis elegans expresses three functional profilins in a tissue-specific manner. Cell Motil. Cytoskeleton 63, 14-28. Abstract Article
Price, M.G., Landsverk, M.L., Barral, J.M., and Epstein, H.F. (2002). Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific function. J. Cell Sci. 115, 4013-4023. Abstract Article
Probst, W.C., Cropper, E.C., Heierhorst, J., Hooper, S.L., Jaffe, H., Vilim, F., Beushausen, S., Kupfermann, I., and Weiss, K.R. (1994). cAMP-dependent phosphorylation of Aplysia twitchin may mediate modulation of muscle contractions by neuropeptide cotransmitters. Proc. Natl. Acad. Sci. U. S. A. 91, 8487-8491. Abstract Article
Puchner, E.M., Alexandrovich, A., Kho, A.L., Hensen, U., Schäfer, L.V, Brandmeier, B., Gräter, F., Grubmüller, H., Gaub, H.E., and Gautel, M. (2008). Mechanoenzymatics of titin kinase. Proc. Natl. Acad. Sci. U. S. A. 105, 13385-13390. Abstract Article
Qadota, H., Mercer, K.B., Miller, R.K., Kaibuchi, K., and Benian, G.M. (2007). Two LIM domain proteins and UNC-96 link UNC-97/pinch to myosin thick filaments in Caenorhabditis elegans muscle. Mol. Biol. Cell 18, 4317-4326. Abstract Article
Qadota, H., McGaha, L.A., Mercer, K.B., Stark, T.J., Ferrara, T.M. and Benian, G.M. (2008a). A novel protein phosphatase is a binding partner for the protein kinase domains of UNC-89 (Obscurin) in Caenorhabditis elegans. Mol. Biol. Cell 19, 2424-2432. Abstract Article
Qadota, H., Blangy, A., Xiong, G. and Benian, G.M. (2008b). The DH-PH region of the giant protein UNC-89 activates RHO-1 GTPase in Caenorhabditis elegans body wall muscle. J. Mol. Biol. 383, 747-752. Abstract Article
Qadota, H., and Benian, G.M. (2010). Molecular structure of sarcomere-to-membrane attachment at M-lines in C. elegans muscle. J. Biomed. Biotechnol. 2010, 864749. Abstract Article
Qadota, H., Miyauchi, T., Nahabedian, J.F., Stirman, J.N., Lu, H., Amano, M., Benian, G.M., and Kaibuchi, K. (2011). PKN-1, a homologue of mammalian PKN, is involved in the regulation of muscle contraction and force transmission in C. elegans. J. Mol. Biol. 407, 222-231. Abstract Article
Qadota, H., Moerman, D.G., and Benian, G.M. (2012). A molecular mechanism for the requirement of PAT-4 (integrin-linked kinase (ILK)) for the localization of UNC-112 (kindlin) to integrin adhesion sites. J. Biol. Chem. 287, 28537-28551. Abstract Article
Qadota, H., Luo, Y., Matsunaga, Y., Park, A.S., Gernert, K.M., and Benian, G.M. (2014). Suppressor mutations suggest a surface on PAT-4 (integrin linked kinase) that interacts with UNC-112 (kindlin). J. Biol. Chem. 289, 14252-14262. Abstract Article
Qadota, H., and Benian, G.M. (2014). An approach for exploring interaction between two proteins in vivo. Fron. Physiol. 5, 162. Abstract
Qadota, H., Mayans, O., Matsunaga, Y., McMurry, J.L., Wilson, K.J., Kwon, G.E., Stanford, R., Deehan, K., Tinley, T.L., Ngwa, M.M., and Benian, G.M. (2016). The SH3 domain of UNC-89 (obscurin) interacts with paramyosin, a coiled-coil protein, in C. elegans muscle. Mol. Biol. Cell 27, 1606-1620. Abstract Article
Rabets, Y., Backholm, M., Dalnoki-Veress, K., and Ryu, W.S. (2014). Direct measurements of drag forces in C. elegans crawling locomotion. Biophys. J. 107, 1980-1987. Abstract Article
Rahmani, P., Rogalski, T., and Moerman, D.G. (2015). The C. elegans UNC-23 protein, a member of the BCL-2-associated athanogene (BAG) family of chaperone regulators, interacts with SHP-1 to regulate cell attachment and maintain hyodermal integrity. Worm 4, e1023496. Abstract Article
Raizen, D.M., Zimmerman, J.E., Maycock, M.H., Ta, U.D., You, Y., Sundaram, M.V., and Pack, A.I. (2008). Lethargus is a Caenorhabditis elegans sleep-like state. Nature 451, 569–572. Abstract Article
Regmi, SG., Rolland, SG. and Conradt, B. (2014). Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging 6, 118-130. Abstract Article
Resnicow, D.I., Deacon, J.C., Warrick, H.M., Spudich, J.A., and Leinwand, L.A. (2010). Functional diversity among a family of human skeletal muscle myosin motors. Proc. Natl. Acad. Sci. U. S. A. 107, 1053-1058. Abstract Article
Restif, C., Ibáñez-Ventoso, C., Vora, M.M., Guo, S., Metaxas, D., and Driscoll, M. (2014). CeleST: Computer vision software for quantitative analysis of C. elegans swim behavior reveals novel features of locomotion. PLoS Comput. Biol. 10, e1003702 Abstract Article
Riddle, D.L., and Brenner, S. (1978). Indirect suppression in C. elegans. Genetics 89, 299-314. Abstract
Rogalski, T.M., Williams, B.D., Mullen, G.P., and Moerman, D.G. (1993). Products of the unc-52 gene in Caenorhabditis elegans are homologous to the core protein of the mammalian basement membrane heparan sulfate proteoglycan. Genes Dev. 7, 1471-1484. Abstract Article
Rogalski, T.M., Mullen, G.P., Gilbert, M.M., Williams, B.D., and Moerman, D.G. (2000). The UNC-112 gene in C. elegans encodes a novel component of cell-matrix adhesion structures required for integrin localization in the muscle cell membrane. J. Cell Biol. 150, 253-264. Abstract Article
Rogalski, T.M., Gilchrist, E.J., Mullen, G.P., and Moerman, D.G. (1995). Mutations in the unc-52 gene responsible for body wall muscle defects in adult Caenorhabditis elegans are located in alternatively spliced exons. Genetics 139, 159-169. Abstract
Rogalski, T.M., Gilbert, M.M., Devenport, D., Norman, K.R., and Moerman, D.G. (2003). DIM-1, a novel immunoglobulin superfamily protein in Caenorhabditis elegans, is necessary for maintaining bodywall muscle integrity. Genetics 163, 905-915. Abstract
Rybakova, I.N., Patel, J.R., and Ervasti, J.M. (2000). The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 150, 1209–1214. Abstract Article
Saksela, K., and Permi, P. (2012). SH3 domain ligand binding: What's the consensus and where's the specificity? FEBS Lett. 586, 2609-2614. Abstract Article
Samarel, A.M. (2005). Costameres, focal adhesions, and cardiomyocyte mechanotransduction. Am. J. Physiol. Heart Circ. Physiol. 289, H2291-H2301. Abstract Article
Schachat, F., Garcea, R.L., and Epstein, H.F. (1978). Myosins exist as homodimers of heavy chains: demonstration with specific antibody purified by nematode mutant myosin affinity chromatography. Cell 15, 405-411. Abstract Article
Schriefer, L.A., and Waterston, R.H. (1989). Phosphorylation of the N-terminal region of Caenorhabditis elegans paramyosin. J. Mol. Biol. 207, 451-454. Abstract Article
Schwechheimer, C. (2004). The COP9 signalosome (CSN): an evolutionary conserved proteolysis regulator in eukaryotic development. Biochem. Biophys. Acta 1695, 45-54. Abstract Article
Ségalat, L., and Anderson, J.E. (2005). Duchenne muscular dystrophy: Stalled at the junction? Eur. J. Hum. Genet. 13, 4–5. Abstract
Shen, X.N., Sznitman, J., Krajacic, P., Lamitina, T., and Arratia, P.E. (2012). Undulatory locomotion of Caenorhabditis elegans on wet surfaces. Biophys. J. 102, 2772-2781. Abstract Article
Siegman, M.J., Funabara, D., Kinoshita, S., Watabe, S., Hartshorne, D.J., and Butler, T.M. (1998). Phosphorylation of a twitchin-related protein controls catch and calcium sensitivity of force production in invertebrate smooth muscle. Proc. Natl. Acad. Sci. U. S. A. 95, 5383-5388. Abstract
Small, T.M., Gernert, K.M., Flaherty, D.B., Mercer, K.B., Borodovsky, M., and Benian, G.M. (2004). Three new isoforms of C. elegans UNC-89 containing MLCK-like protein kinase domains. J. Mol. Biol. 342, 91-108. Abstract Article
Spooner, P.M., Bonner, J., Maricq, A.V., Benian, G.M., and Norman, K.R. (2012). Large isoforms of UNC-89 (obscurin) are required for muscle cell architecture and optimal calcium release in Caenorhabditis elegans. PLoS One 7, e40182. Abstract Article
Swierczek, N.A., Giles, A.C., Rankin, C.H., and Kerr, R.A. (2011). High-throughput behavioral analysis in C. elegans. Nat. Methods 8, 592-598. Abstract Article
Takeda, K., Watanabe, C., Qadota, H., Hanazawa, M., and Sugimoto, A. (2008). Efficient production of monoclonal antibodies recognizing specific structures in Caenorhabditis elegans embryos using an antigen subtraction method. Genes Cells 13, 653-665. Abstract Article
Taylor, R.C. and Dillin, A. (2011). Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3, a004440 Abstract
Thompson, O., Edgley, M., Strasbourger, P., Flibotte, S., Ewing, B., Adair, R., Au, V., Chaudhry, I., Fernando, L., Hutter, H., et al. (2013). The million mutation project: a new approach to genetics in Caenorhabditis elegans. Genome Res. 23, 1749-1762. Abstract Article
Turek, M., and Bringmann, H. (2014). Gene expression changes of Caenorhabditis elegans larvae during molting and sleep-like lethargus. PloS One 9, e113269. Abstract Article
Turner, P.R., Westwood, T., Regen, C.M., and Steinhardt, R.A. (1988). Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 335, 735–738. Abstract Article
van, Oosten-Hawle, P. and Morimoto, RI. (2014) Organismal proteostasis: role of cell-nonautonomous regulation and transcellular chaperone signaling. Genes and Dev. 28, 1533–1543. Abstract Article
Varkuti, B.H., Yang, Z., Kintses, B., Erdelyi, P., Bardos-Nagy, I., Kovacs, A.L., Hari, P., Kellermayer, M.,Vellai, T., and Malnasi-Csizmadia, A. (2012). A novel actin binding site of myosin required for effective muscle contraction. Nat. Struct. Mol. Biol. 19, 299-307. Abstract Article
Venolia, L., and Waterston, R.H. (1990). The unc-45 gene of Caenorhabditis elegans is an essential muscle-affecting gene with maternal expression. Genetics 126, 345-353. Abstract
Venolia, L., Ao, W., Kim, S., Kim, C., and Pilgrim, D. (1999). unc-45 gene of Caenorhabditis elegans encodes a muscle-specific tetratricopeptide repeat-containing protein. Cell Motil. Cytoskeleton 42, 163-177. Abstract
von Castelmur, E., Strümpfer, J., Franke, B., Bogomolovas, J., Barbieri, S., Qadota, H., Konarev, P,V., Svergun, D.I., Labeit, S., Benian, G.M., et al. (2012). Identification of an N-terminal inhibitory extension as the primary mechanosensory regulator of twitchin kinase. Proc. Natl. Acad. Sci. U. S. A. 109, 13608-13613. Abstract Article
Wang, Z.W., Saifee, O., Nonet, M.L. and Salkoff, L. (2001). SLO-1 potassium channels control quantal content of neurotransmitter release at the C. elegans neuromuscular junction. Neuron 32, 867-81. Abstract Article
Warner, A., Qadota, H., Benian, G.M., Vogl, A.W., and Moerman, D.G. (2011). The C. elegans paxillin orthologue, PXL-1, is required for pharyngeal muscle contraction and for viability. Mol. Biol. Cell 22, 2551-2563. Abstract Article
Warner, A., Xiong, G., Qadota, H., Rogalski, T., Vogl, A.W., Moerman, D.G., and Benian, G.M. (2013). CPNA-1, a copine domain protein, is located at integrin adhesion sites and is required for myofilament stability in C. elegans. Mol. Biol. Cell 24, 601-616. Abstract Article
Waterston, R.H., Fishpool, R.M., and Brenner, S. (1977). Mutants affecting paramyosin in Caenorhabditis elegans. J. Mol. Biol. 117, 679-697. Abstract Article
Waterston, R.H., Thomson, J.N., and Brenner, S. (1980). Mutants with altered muscle structure in C. elegans. Dev. Biol. 77, 271-302. Abstract Article
Waterston, R.H. (1988). Muscle. In The Nematode Caenorhabditis elegans, W.B. Wood, ed. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press) pp. 281-335. xs
Waterston, R.H. (1989). The minor myosin heavy chain, MHC A, of Caenorhabditis elegans is necessary for the initiation of thick filament assembly. EMBO J. 8, 3429-3436. Abstract
Williams, B.D., and Waterston, R.H. (1994). Genes critical for muscle development and function in Caenorhabditis elegans identified through lethal mutations. J. Cell Biol. 124, 475-490. Abstract Article
Wilson, K.J., Qadota, H., Mains, P.E., and Benian, G.M. (2012). UNC-89 (obscurin) binds to MEL-26, a BTB domain protein, and affects the function of MEI-1 (katanin) in striated muscle of C. elegans. Mol. Biol. Cell 23, 2623-2634. Abstract Article
Xu, L., Wei, Y., Reboul, J., Vaglio, P., Shin, T.H., Vidal, M., Elledge, S.J., and Harper, J.W. (2003). BTB proteins are substrate-specific adaptors in an SCF-lke modular ubiqutin ligase containing CUL-3. Nature 425, 316-321. Abstract Article
Xiong, G., Qadota, H., Mercer, K.B., McGaha, L.A., Oberhauser, A.F., and Benian, G.M. (2009). A LIM-9 (FHL)/SCPL-1 (SCP) complex interacts with the C-terminal protein kinase regions of UNC-89 (obscurin) in C. elegans muscle. J. Mol. Biol. 386, 976-988. Abstract Article
Yamashiro, S., Cox, E.A., Baillie, D.L., Hardin, J.D., and Ono, S. (2008). Sarcomeric actin organization is synergistically promoted by tropomodulin, ADF/cofilin, AIP1 and profilin in C. elegans. J. Cell Sci. 121, 3867-3877. Abstract Article
Young, P., Ehler, E., and Gautel, M. (2001). Obscurin, a giant sarcomeric Rho guanine nucleotide exchange factor protein involved in sarcomere assembly. J. Cell Biol. 154, 123-136. Abstract Article
Yu, X., Ma, J., Lin, F., Zhao, W., Fu, X., and Zhao, Z.J. (2012) Myotubularin family phosphatase ceMTM3 is required for muscle maintenance by preventing excessive autophagy in Caenorhabditis elegans. BMC Cell Biol. 13, 28. Abstract Article
Yuan, L., Fairchild, M.J., Perkins, A.D., and Tanentzapf, G. (2010). Analysis of integrin turnover in fly myotendinous junctions. J. Cell Sci. 123, 939-946. Abstract Article
Zaidel-Bar, R., and Geiger, B. (2010). The switchable integrin adhesome. J. Cell Sci. 123, 1385-1388. Abstract Article
Zaidel-Bar, R., Miller, S., Kaminsky, R., and Broday, L. (2010). Molting-specific downregulation of C. elegans body-wall muscle attachment sites: the role of RNF-5 E3 ligase. Biochem. Biophys. Res. Commun. 395, 509–514. Abstract Article
Zengel, J.M., and Epstein, H.F. (1980). Identification of genetic elements associated with muscle structure in the nematode Caenorhabditis elegans. Cell Motil. 1, 73-97. Abstract Article
*Edited by Michel Labouesse. Last revised June 25, 2016. Published in its final form April 13, 2017. This chapter should be cited as: Gieseler K., Qadota H., and Benian G. M. Development, structure, and maintenance of C. elegans body wall muscle. (April 13, 2017), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.81.2, http://www.wormbook.org.
Copyright: © 2016 Kathrin Gieseler, Hiroshi Qadota, and Guy M. Benian. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed:. E-mail: pathgb@emory.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.