Embryo series courtesy of Einhard Schierenberg
Embryo series courtesy of Einhard SchierenbergTable of Contents
Abstract
Receptor Tyrosine Kinase (RTK)-Ras-Extracellular signal-regulated kinase (ERK) signaling pathways control many aspects of C. elegans development and behavior. Studies in C. elegans helped elucidate the basic framework of the RTK-Ras-ERK pathway and continue to provide insights into its complex regulation, its biological roles, how it elicits cell-type appropriate responses, and how it interacts with other signaling pathways to do so. C. elegans studies have also revealed biological contexts in which alternative RTK- or Ras-dependent pathways are used instead of the canonical pathway.
The small GTPase Ras is a central player in multiple different signaling pathways, but one of the most important and well conserved of these is the RTK-Ras-ERK pathway (Malumbres and Barbacid, 2002; Yoon and Seger, 2006; Karnoub and Weinberg, 2008; Lemmon and Schlessinger, 2010; Udell et al., 2011) (Figure 1A). RTK-Ras-ERK signaling is used repeatedly during animal development to control many different biological processes, and mutations affecting RTK-Ras-ERK signaling cause many human syndromes and diseases, including Noonan syndrome, Costello syndrome and many types of cancers (Karnoub and Weinberg, 2008; Tidyman and Rauen, 2009).
|
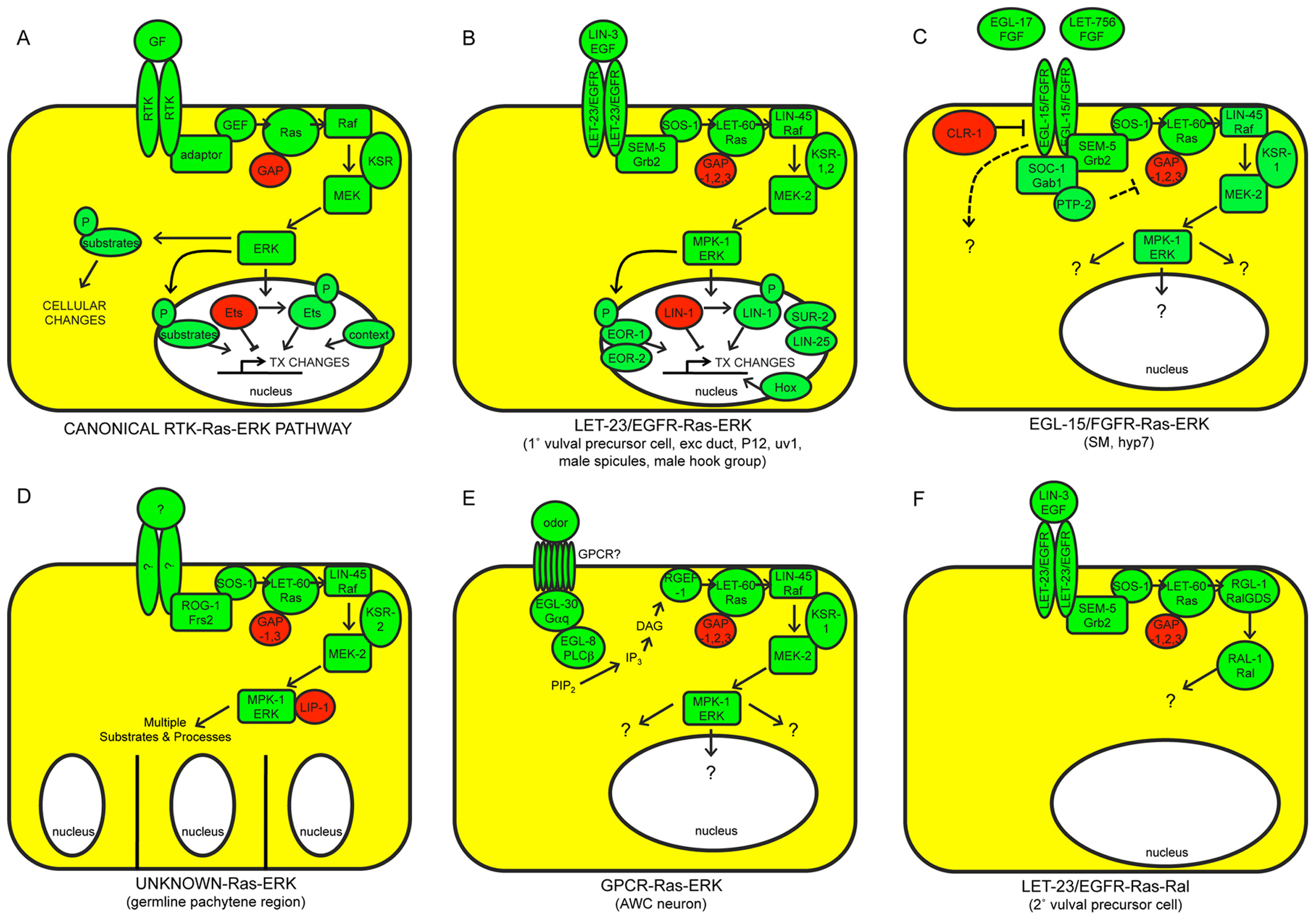
Figure 1. The canonical RTK-Ras-ERK signaling pathway and alternative Ras-dependent pathways in C. elegans. A) The canonical pathway. Positively acting factors are shown in green; negatively acting factors are in red. Only the most widely required and conserved factors are shown. GF, growth factor. TX, transcription. B) LET-23/EGFR and C) EGL-15/FGFR both act in part through the canonical pathway, but they are activated by different ligands and control different sets of developmental processes. Included in B) are nuclear proteins, including two MPK-1/ERK substrates (LIN-1/ETS and EOR-1), that are jointly important for most LET-23-dependent somatic cell fates. Downstream targets of EGL-15-Ras-ERK signaling are not yet known. D) Ras-ERK functions to promote meiotic pachytene progression and various other aspects of germline development; the receptors that activate Ras-ERK for these functions are not currently known. MPK-1/ERK utilizes more than 30 different substrates in the germline. E) A G-protein coupled receptor (GPCR) may activate Ras-ERK signaling in some contexts. F) LET-60/Ras can signal through the alternative Ral pathway. See text for additional details and references.
This review summarizes work on RTK-Ras-ERK signaling in C. elegans. In an attempt to make this review accessible to a broad audience, I use the more familiar names of the mammalian proteins in conjunction with, or often in place of, the C. elegans nomenclature. For example, I refer to the C. elegans Ras protein as LET-60/Ras or simply Ras. See Figure 1 and Table 1 for corresponding names of core signaling proteins in C. elegans vs. mammals.
Table 1. Core components and selected regulators and targets of the C. elegans RTK-Ras-ERK signaling pathway. Core components are shown in black, positive regulators or targets are shown in green, and negative regulators or targets are shown in red.
| Gene Product | Mammalian Relative(s) | Molecular Function | Ras-related Phenotypes | Reference(s) |
| ARK-1(Ack-Related Kinase) | Ack | Cytoplasmic tyrosine kinase | ∼WT. Muv in combination with sli-1, others. | (Hopper et al., 2000) |
| CDF-1 (Cation Diffusion Facilitator) | ZnT1 | Zinc transporter | ∼WT. Suppresses let-60 ras(gf) Muv. | (Bruinsma et al., 2002; Jakubowski and Kornfeld, 1999) |
| CGR-1 (CRAL/TRIO and GOLD domain suppressor of activated Ras) | SPF/TAP-1 | CRAL/TRIO and GOLD domains | ∼WT. Suppresses let-60(gf) Muv. | (Goldstein et al., 2006) |
| CLR-1 (CLeaR) | LAR family | Receptor Tyrosine Phosphatase | Lethal fluid accumulation, Suppressed by egl-15(rf) | (Kokel et al., 1998) |
| CNK-1 (Connector/eNhancer of Ksr) | CNK1,2,3 | Scaffold protein | ∼WT. Suppresses let-60 ras(gf) Muv. Enhances Vul and rod-like lethal phenotypes of lin-45, others. | (Rocheleau et al., 2005) |
| DAB-1 (DisABled homolog) | DAB2 | Adaptor. Required for EGL-17 secretion. | Abnormal sex myoblast positions. | (Kamikura and Cooper, 2003) |
| DEP-1 (Density Enhanced Phosphatase) | Dep1 | Receptor Tyrosine Phosphatase | ∼WT. Muv and excretory duct duplication in combination with lip-1, others. | (Berset et al., 2005) |
| DPY-23 (DumPY) | AP-50 | Clathrin Adaptor subunit | ∼WT. Muv in combination with gap-1. | (Yoo et al., 2004) |
| EGL-15 (EGg Laying defects) | FGFR | Receptor Tyrosine Kinase | Larval lethal. Abnormal sex myoblast positions. Axon guidance defects. | (Bulow et al., 2004; DeVore et al., 1995; Goodman et al., 2003; Stern and Horvitz 1991) |
| EGL-17 (EGg-Laying defects) | FGF8 | Ligand for EGL-15 RTK | Abnormal sex myoblast positions. | (Burdine et al., 1998; Stern and Horvitz 1991) |
| EOR-1 (Egl-1 suppressor/diO uptake defective/Raf enhancer) | PLZF | BTB/Zinc finger protein, probable transcriptional regulator and MPK-1 substrate | Partial rod-like lethal and P12→P11 fate changes. Strong rod-like lethal in combination with sur-2, lin-25 or lin-1. Reduced age-dependent ubiquitation. | (Howard and Sundaram, 2002; Howell et al., 2010; Liu et al., 2011; Rocheleau et al., 2002) |
| EOR-2 (Egl-1 suppressor/diO uptake defective/Raf enhancer) | NP_079001.2 | Novel nuclear protein, functions with EOR-1 | Partial rod-like lethal and P12→P11 fate changes. Strong rod-like lethal in combination with sur-2, lin-25 or lin-1. Reduced age-dependent ubiquitation. | (Howard and Sundaram, 2002; Howell et al., 2010; Liu et al., 2011; Rocheleau et al., 2002) |
| EPS-8 (Egfr Protein Substrate 8) | EPS8 | EGFR substrate and regulator of EGFR trafficking | ∼WT. Enhances lin-3(rf) or let-23(rf) Vul. | (Stetak et al., 2006) |
| GAP-1 (GTPase Activating Protein) | GAP-1 | Ras GAP | ∼WT. Muv and Clr in combination with gap-3. Excretory duct duplication in combination with gap-2. | (Hajnal et al., 1997; Stetak et al., 2008) |
| GAP-2 (GTPase Activating Protein) | synGAP | Ras GAP | ∼WT. Excretory duct duplication in combination with gap-1. | (Hayashizaki et al., 1998; Stetak et al., 2008) |
| GAP-3 (GTPase Activating Protein) | p120GAP | Ras GAP | Premature pachytene exit. Muv and Clr in combination with gap-1. | (Stetak et al., 2008) |
| GCK-1 (Germinal Center Kinase) | GCK III | Serine/Threonine kinase, binds MPK-1 | Increased germline apoptosis, oocyte defects | (Schouest et al., 2009) |
| GLA-3 (GermLine Apoptosis) | Tis11 | CCCH zinc finger protein, binds MPK-1 | Increased germline apoptosis, oocyte defects | (Kritikou et al., 2006) |
| GPA-5 (G-Protein, Alpha subunit) | GNAZ | Gα | Increased chemotaxis. Suppresses let-60(dn) Vul. | (Battu et al, 2003) |
| KSR-1 (Kinase Suppressor of Ras) | KSR1, 2 | Raf-related MEK-binding protein, scaffold for Raf/MEK/ERK | Abnormal sex myoblast positions. Suppresses let-60 ras(gf) Muv. Rod-like lethal and Vul in combination with ksr-2, others. | (Kornfeld et al., 1995b; Sundaram and Han, 1995) |
| KSR-2 (Kinase Suppressor of Ras) | KSR1, 2 | Raf-related MEK-binding protein, scaffold for Raf/MEK/ERK | Sterile. Rod-like lethal and Vul in combination with ksr-1. | (Ohmachi et al., 2002) |
| LET-23 (LEThal) | EGFR | Receptor Tyrosine Kinase | Rod-like larval lethal, Vul, sterile, etc. | (Aroian et al., 1990) |
| LET-60 (LEThal) | K-Ras | Small GTPase | Rod-like larval lethal, Vul, sterile, etc. | (Beitel et al., 1990; Han et al., 1990; Han and Sternberg, 1990) |
| LET-92 (LEThal) | PPP2CB | Catalytic subunit of Protein Phosphatase 2A | Larval lethal. Dominantly enhances let-60(dn) Vul | (Kao et al., 2004) |
| LET-756 (LEThal) | FGF9 | Ligand for EGL-15 RTK | Larval lethal. Axon guidance defects. | (Bulow et al., 2004; Popovici et al., 2004; Roubin et al., 1999) |
| LIN-1 (abnormal cell LINeage) | Elk1 | Ets domain transcription factor. Substrate of MPK-1. | Muv, excretory duct duplication, etc. Rod-like lethal in combination with eor-1 or eor-2. | (Beitel et al., 1995; Jacobs et al., 1998; Howard and Sundaram, 2002) |
| LIN-2 (abnormal cell LINeage) | CASK | Membrane-associated guanylate kinase, required for basal localization of LET-23 | Vul | (Hoskins et al., 1996; Kaech et al., 1998) |
| LIN-3 (abnormal cell LINeage) | EGF | Ligand for LET-23 RTK | Rod-like larval lethal, Vul, Sterile, etc. | (Hill and Sternberg, 1992; Dutt et al., 2004) |
| LIN-7 (abnormal cell LINeage) | Lin-7 | PDZ and PTB-domain protein, required for basal localization of LET-23 | Vul | (Kaech et al., 1998; Simske et al., 1996) |
| LIN-10 (abnormal cell LINeage) | Mint1, 2, 3 | PDZ protein, required for basal localization of LET-23 | Vul | (Kaech et al., 1998; Whitfield et al., 1999) |
| LIN-15A and LIN-15B (abnormal cell LINeage) | - | Adjacent SynMuvA and SynMuv B genes encoding nuclear proteins that redundantly inhibit lin-3 expression | double mutant Muv, excretory duct duplication, etc. | (Ferguson and Horvitz, 1989; Huang et al., 1994; Cui et al., 2006) |
| LIN-25 (abnormal cell LINeage) | - | Novel nuclear protein, functions with SUR-2 | Vul. Rod-like lethal in combination with eor-1, others. | (Nilsson et al., 1998; Nilsson et al., 2000; Tuck and Greenwald, 1995; Howard and Sundaram, 2002) |
| LIN-31 (abnormal cell LINeage) | FoxB2 | Winged helix transcription factor. Target of MPK-1. | Mixed Vul and Muv | (Miller et al., 1993; Tan et al., 1998) |
| LIN-45 (abnormal cell LINeage) | B-Raf | Serine/Threonine Kinase. Binds Ras-GTP, phosphorylates MEK. | Sterile Vul (maternal rescue of rod-like lethal), etc. | (Han et al., 1993; Hsu et al., 2002) |
| LIP-1 (Lateral signal Induced Phosphatase) | MKP1 | MAPK phosphatase | Partial sterile. Muv in combination with gap-1, others. | (Berset et al., 2001; Hajnal and Berset, 2002) |
| LPR-1 (LiPocalin Related) | lipocalin family | secreted protein | rod-like lethal | (Stone et al., 2009) |
| LRP-1 (low-density Lipoprotein Receptor-related Protein) | LRP1 | Lipoprotein Receptor-related protein. Required for EGL-17 secretion. | Abnormal sex myoblast positions. | (Kamikura and Cooper, 2003) |
| LST-1 (Lateral Signaling Target) | - | Novel MPK-1 binding protein | WT. Muv in combination with gap-1. | (Yoo et al., 2004) |
| LST-2 (Lateral Signaling Target) | Zinc finger FYVE domain containing protein 28 | FYVE domain protein | WT. Muv in combination with gap-1. | (Yoo et al., 2004) |
| LST-3 (Lateral Signaling Target) | CCAR1 | SAF-A/B, Acinus and PIAS domain protein | WT. Muv in combination with gap-1. | (Yoo et al., 2004) |
| LST-4 (Lateral Signaling Target) | NM_153271 | Sorting nexin, may promote LET-23 degradation | WT. Muv in combination with gap-1. | (Yoo et al., 2004) |
| MEK-2 (Map kinase kinase or Erk Kinase) | MEK1, 2 | Dual Specificity kinase, phosphorylates ERK | Sterile Vul (maternal rescue of rod-like lethal), etc. | (Kornfeld et al., 1995a; Wu et al, 1995) |
| MPK-1/SUR-1 (MaP Kinase/ SUppressor of Ras) | ERK1, 2 | Serine/Threonine kinase | Sterile Vul (maternal rescue of rod-like lethal), etc. | (Lackner and Kim, 1998; Lackner et al., 1994; Wu and Han, 1994) |
| PAR-1 (abnormal embryonic PARtitioning of cytoplasm) | MARK2/C-TAK1 | Serine/Threonine kinase | Weak Muv. Reverses sur-6 suppressor phenotype. | (Kao et al., 2004; Yoder et al., 2004) |
| PTP-2 (Protein Tyrosine Phosphatase) | Shp-2 | Tyrosine phosphatase | Sterile and maternal-effect lethal. Suppressed by let-60 ras(gf). Suppresses clr-1. | (Gutch et al., 1998; Schutzman et al., 2001) |
| RAB-7 (RAB GTPase) | Rab7 | Rab GTPase | WT. Muv in combination with ark-1 or sli-1. | (Skorobogata and Rocheleau, 2012) |
| RGEF-1 (Ras Guanine nucleotide Exchange Factor) | RasGRP | Guanine Nucleoide Exchange Factor | Chemotaxis-defective | (Chen et al., 2011) |
| ROM-1 (RhOMboid-related) | RHBDL2 | 7-pass transmembrane serine protease | WT. Suppresses let-60 ras(gf) Muv. | (Dutt et al., 2004) |
| SEM-5 (SEx Muscle abnormality) | Grb2 | RTK-binding adaptor | Rod-like larval lethal, Vul, etc. | (Clark et al., 1992) |
| SLI-1 (Suppressor of LIneage defect) | Cbl | E3 ubiquitin ligase, Involved in LET-23 endocytosis/degradation | WT. Muv in combination with unc-101, others. | (Jongeward et al., 1995; Yoon et al., 1995) |
| SOC-1 (Suppressor Of Clr-1) | Gab1 | RTK-binding adaptor, Promotes EGL-15 signaling | Scrawny. Suppresses clr-1 lethality. | (Schutzman et al., 2001) |
| SOC-2/SUR-8 (SUppressor of Ras, Suppressor Of Clr-1) | Shoc2/Sur-8 | Leucine-rich repeat protein, binds Ras and Raf | Scrawny. Suppresses let-60 ras(gf) Muv and clr-1 lethality. Enhances Vul and rod-like lethal phenotypes of lin-45 hypomorphs, others. | (Selfors et al., 1998; Sieburth et al., 1998) |
| SOS-1/LET-341 (Son Of Sevenless/ LEThal) | Sos-1 | Guanine Nucleotide Exchange Factor | Rod-like larval lethal, Vul, Sterile, etc. | (Chang et al., 2000) |
| SRA-13 (Serpentine Receptor, A class) | - | G-protein coupled receptor | Increased chemotaxis. Suppresses let-60(dn) Vul. | (Battu et al., 2003) |
| SUR-2 (SUppressor of Ras) | Med23/Sur-2 | Mediator subunit | Vul. Rod-like lethal in combination with eor-1, others. | (Singh and Han, 1995; Howard and Sundaram, 2002) |
| SUR-5 (SUppressor of Ras) | NM_023928 | Acetyl coenzyme A synthetase | WT. Suppresses let-60(dn) Vul. | (Gu et al., 1998) |
| SUR-6 (SUppressor of Ras) | PPP2R2A | PR55/B Regulatory subunit of Protein Phosphatase 2A | Suppresses let-60 ras(gf) Muv. Enhances Vul and rod-like lethal phenotypes of lin-45 hypomorphs, others. | (Kao et al., 2004; Sieburth et al., 1999) |
| SUR-7 (SUppressor of Ras) | - | Zinc transporter | Suppresses let-60 ras(gf) Muv. | (Yoder et al., 2004) |
| SYNMUV GENES (SYNthetic MUltiVulva) | Rb, E2F, NuRD, SETDB1, HP1, Tip60/NuA4, etc. | inhibitors of lin-3 gene expression | Muv in combination with appropriate other SynMuv mutations | (Ferguson and Horvitz, 1989; Cui et al., 2006; Fay and Yochem, 2007) |
| UNC-101 (UNCoordinated) | AP-47 | Clathrin adaptor subunit | ∼WT. Muv in combination with sli-1, others. | (Lee et al., 1994) |
A general model for the canonical RTK-Ras-ERK signaling pathway is shown in Figure 1A, and canonical pathways in C. elegans are shown in Figures 1B-C. Upon growth factor binding, an RTK dimerizes and autophosphorylates its C-terminal region (Lemmon and Schlessinger, 2010). The resulting phospho-tyrosine residues serve as docking sites for adaptor proteins such as Grb2. These adaptors recruit the Guanine Nucleotide Exchange Factor (GEF) Sos to activate the small GTPase Ras (Karnoub and Weinberg, 2008). Ras-GTP then binds to Raf and promotes its stable association with the plasma membrane and/or endomembranes, where other events then activate Raf kinase activity (Udell et al., 2011). The scaffold protein KSR assists in Raf activation, but also promotes further signal transmission by bringing together different components of the Raf-MEK-ERK kinase cascade (Udell et al., 2011). Raf phosphorylates and activates MEK, MEK phosphorylates and activates ERK, and ERK then phosphorylates and either activates or inactivates various substrates, depending on the cell-type and context (Yoon and Seger, 2006). In many cases, ERK phosphorylates nuclear transcription factors such as Ets domain proteins, thus leading to changes in gene expression. ERK can also phosphorylate a broad array of other, non-transcription factor substrates to affect other cell biological processes.
This model draws on a large body of data from multiple systems, including C. elegans, Drosophila and mammalian cells. C. elegans genetics has been particularly powerful for identifying the gene products required for particular signaling events and determining their order of action; for example, studies in C. elegans were among the first to show that Ras acts downstream of RTKs (Aroian et al., 1990) and upstream of Raf (Han et al., 1993), and to identify key pathway components such as SEM-5/Grb2 (Clark et al., 1992) and KSR (Kornfeld et al., 1995b; Sundaram and Han, 1995). In some cases, physical interactions and/or phosphorylation events have been demonstrated for the worm proteins, but in other cases such interactions are inferred based on biochemical studies of related proteins in mammalian cells. The canonical RTK-Ras-ERK pathway (and much of its regulation) appears highly conserved between C. elegans and mammals (Figure 1A-C; Table 1).
The C. elegans genome contains 33 predicted RTKs, with about half of these belonging to two nematode-specific RTK families (Popovici et al., 1999; see Genomic overview of protein kinases). Of the RTKs that have been characterized, only the Epidermal Growth Factor (EGF) receptor relative LET-23 and the Fibroblast Growth Factor (FGF) receptor relative EGL-15 clearly signal through LET-60/Ras (Aroian et al., 1990; DeVore et al., 1995), and signaling by LET-23 and EGL-15 can account for most (but not all) of the LET-60/Ras-dependent processes described below. The insulin-like RTK DAF-2 may also signal in part through LET-60/Ras (Nanji et al., 2005), but acts primarily through a separate PI3-kinase-Akt pathway (see Dauer). The ephrin RTK VAB-1 negatively regulates MPK-1/ERK activation (Miller et al., 2003). Several RTKs act in other characterized pathways; for example, VAB-1 acts upstream of PLC-3/phospholipase C-gamma (PLCγ) and inositol 1,4,5-triphosphate (IP3) (Yin et al., 2004), SCD-2/ALK acts upstream of the SMA-5 MAP kinase (Reiner et al., 2008), and LIN-18/Ryk and CAM-1/Ror act in Wnt signaling pathways (Inoue et al., 2004; Green et al., 2007, Green et al., 2008). Although mutations are now available for most C. elegans RTKs (www.wormbase.org), the biological roles of many of these RTKs remain unknown.
In this review, I focus on LET-23/EGFR and EGL-15/FGFR, the two RTKs that signal widely (though not exclusively) through Ras-ERK. LET-23 has one known ligand, LIN-3, the sole EGF-related protein in C. elegans (Hill and Sternberg, 1992) (Figure 1B). EGL-15 has two different FGF-related ligands, EGL-17 and LET-756, that are used in different biological contexts (Burdine et al., 1997; Roubin et al., 1999; Birnbaum et al., 2005; Huang and Stern, 2005) (Figure 1C). EGL-17 and LET-756 are most related to the vertebrate FGF8 and FGF9 families, respectively (Popovici et al., 2004; Birnbaum et al., 2005).
As in other organisms, several alternative signaling pathways in C. elegans utilize only part of the core RTK-Ras-ERK pathway.
Alternative receptors: Unknown receptors, potentially not RTK-related, can stimulate Ras-ERK signaling (Figure 1D, 1E). In particular, there is evidence for G-protein-coupled receptor (GPCR)-dependent activation of Ras in neurons (Chen et al., 2011) (Figure 1E).
Alternative Ras targets: LET-60/Ras can act independently of Raf-MEK-ERK to stimulate an alternative pathway involving RGL-1 (related to mammalian Ral guanine nucleotide dissociation stimulator or RalGDS) and the RAL-1 GTPase (Zand et al., 2011) (Figure 1F). Evidence for LET-60/Ras-dependent regulation of other well-known mammalian Ras effectors, such as PI-3 kinase, is so far lacking in C. elegans, but LET-60/Ras can bind to other candidate effectors such as AFD-1/AF-6 (Watari et al., 1998) and PLC-1/PLCε (Shibatohge et al., 1998). Whether Ras acts through Raf-MEK-ERK or through alternative pathways has not yet been established for all known Ras-dependent events (particularly those involving the EGL-15 RTK). There is at least one situation in which the Raf-MEK-ERK cascade appears to act at least partly independently of LET-60/Ras (Nicholas and Hodgkin, 2004; McMullan et al., 2012).
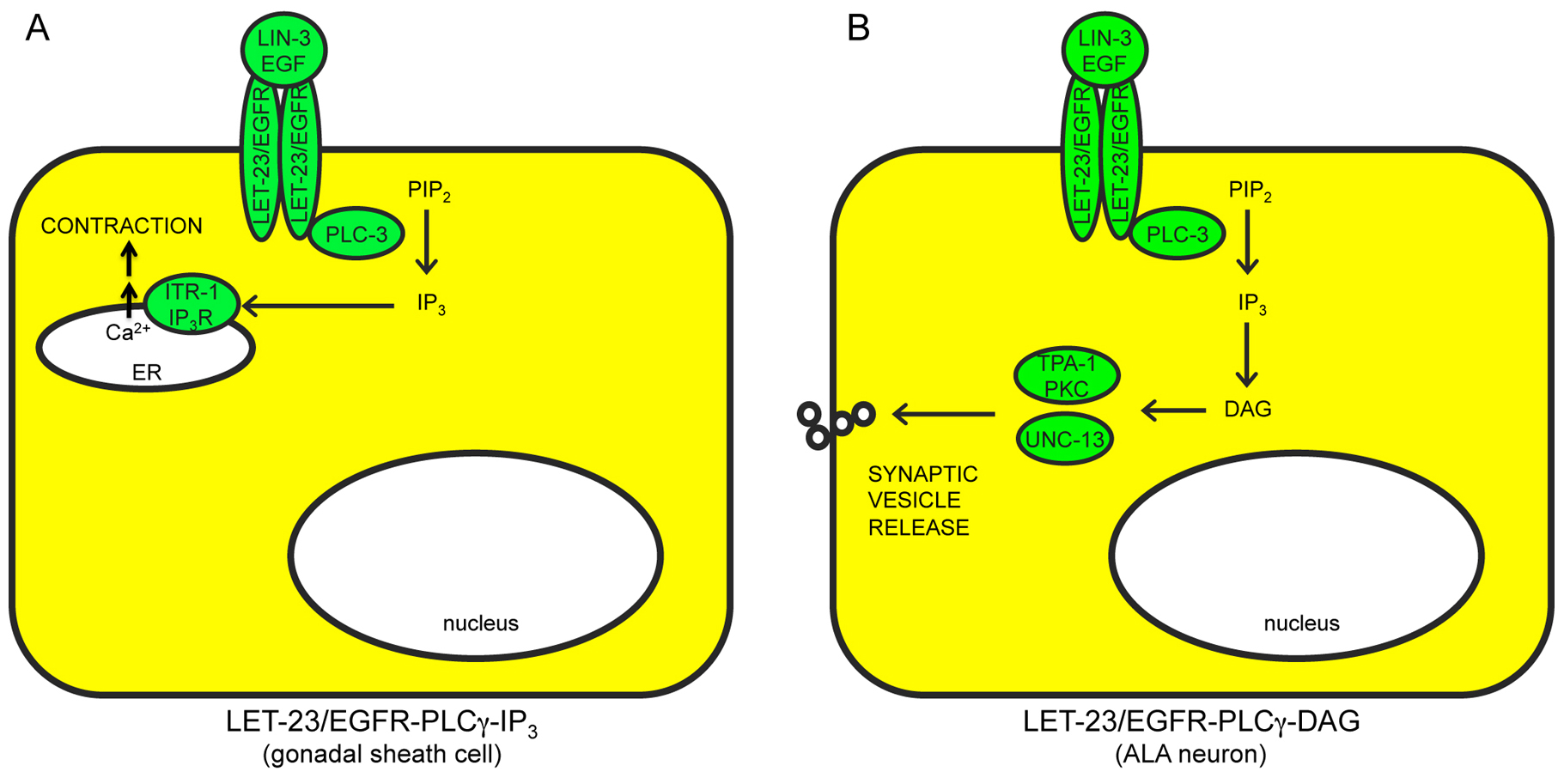
Alternative RTK targets: LET-23/EGFR can stimulate other, Ras-independent pathways such as PLC-3/PLCγ and IP3-dependent release of intracellular calcium stores (Clandinin et al., 1998; Yin et al., 2004) (Figure 2A), or PLC-3 and diacylglycerol (DAG)-dependent release of synaptic vesicles (Van Buskirk and Sternberg, 2007) (Figure 2B). EGL-15/FGFR also may act through additional, unknown pathways (Figure 1C), since constitutive LET-60/Ras cannot bypass all egl-15 requirements (DeVore et al., 1995; Sundaram et al., 1996; Schutzman et al., 2001). Possible direct substrates of EGL-15 kinase activity include the planar polarity protein VANG-1 (Hoffmann et al., 2010) and the cell adhesion molecule LAD-1/L1CAM (Chen et al., 2001). Furthermore, the extracellular domain of EGL-15A can act in a kinase-independent manner to affect axon positions (Bulow et al., 2004). LET-23 appears to signal through Ras-dependent vs. Ras-independent pathways in different cell types, with Ras-dependent signaling promoting transcriptional changes (Figure 1B), and Ras-independent signaling promoting other, non-transcriptional responses (Figure 2A, B). EGL-15 may signal through both Ras-dependent and Ras-independent pathways within the same cell (Figure 1C).
|
Figure 2. Ras-independent pathways downstream of LET-23/EGFR. A) LET-23-PLCγ-IP3 stimulates calcium release from intracellular stores. ER, endoplasmic reticulum. B) LET-23-PLCγ-DAG stimulates synaptic vesicle release. Positively acting factors are shown in green. See text for details and references.
Biological roles of both the canonical pathway and known alternative pathways are summarized in Figure 3 and discussed below.
|
Figure 3. Biological roles of canonical RTK-Ras-ERK signaling and alternative pathways. A) Cell types in the hermaphrodite that require LET-23/EGFR, EGL-15/FGFR and/or LET-60/Ras-dependent signaling are colored according to the type of pathway involved. Not all biological roles are included here due to limited information about the relevant pathway or site of action. Note that many signaling events actually occur during embryogenesis or during larval development, but the locations of relevant cells (or their descendants) in the adult are shown here. B) Photomicrographs of selected mutant phenotypes observed in Ras pathway mutants. Left column, wild type; Right column, mutants. See text for details and references. Scale bar, 10 microns.
Ras signaling is not required for cellular viability or proliferation in C. elegans; in a mosaic analysis, loss of let-60/Ras from large portions of the cell lineage was still compatible with continued cell division and organismal viability, albeit with specific developmental alterations (Yochem et al., 1997). Similarly, increased Ras activity does not promote somatic cell proliferation in C. elegans. The absence of cell-cycle related phenotypes has facilitated studies of Ras requirements in other specific developmental, behavioral and physiological processes.
A let-60/Ras reporter is widely expressed during development (Dent and Han, 1998), and mutations affecting Ras signaling can cause many different phenotypes, including lethality, reflecting the many biological roles of Ras in C. elegans (Figure 3; Table 1). Null mutations in lin-3/EGF, let-23/EGFR, sem-5/Grb2, sos-1/Sos, and let-60/Ras cause “rod-like” L1 larval lethality due to defects in excretory duct cell development (Ferguson and Horvitz, 1985; Yochem et al., 1997; Abdus-Saboor et al., 2011) (Figure 3B). Null mutations in let-756/FGF and egl-15/FGFR cause a distinct (non-rod-like) L1 lethality due to hypodermal defects (DeVore et al., 1995; Roubin et al., 1999; Bulow et al., 2004). Null mutants for lin-45/Raf, mek-2/MEK or mpk-1/ERK are maternally-rescued for earlier essential functions and display an adult sterile phenotype due to germline defects (Church et al., 1995; Lackner and Kim, 1998; Hsu et al., 2002; Lee et al., 2007) (Figure 3B). ksr-2 null mutants show a similar sterile phenotype (Ohmachi et al., 2002). Because of the lethal or sterile phenotypes of null mutants, studies of other biological processes often require the use of conditional or non-null alleles or RNAi.
Excretory duct cell fate and differentiation: LIN-3-LET-23 signaling via Ras-ERK promotes multiple aspects of excretory duct cell development, including excretory duct vs. excretory pore fate specification and other aspects of duct tube morphogenesis and differentiation (Yochem et al., 1997; Abdus-Saboor et al., 2011). The excretory duct cell is a tubular component of the worm's renal-like organ and is required for osmoregulation (Nelson et al., 1983; Nelson and Riddle, 1984). Therefore, let-60/Ras loss-of-function mutants, which lack the excretory duct cell, die as “rod-like” L1 larvae with a turgid, fluid-filled appearance (Figure 3B). let-60/Ras gain-of-function mutants (which are viable) have two fused excretory duct cells (Yochem et al., 1997; Abdus-Saboor et al., 2011). The excretory duct cell was the sole lethal focus identified in a mosaic analysis of let-60 ras (Yochem et al., 1997).
A mosaic analysis of let-23/EGFR identified two lethal foci, one corresponding to the ABpl lineage (which generates the excretory duct) and another corresponding to the ABa lineage (Koga and Ohshima, 1995). Therefore, let-23 appears to have an additional, as yet unknown, essential role.
Hypodermal fluid homeostasis: LET-756-EGL-15 signaling via Ras-ERK also appears to have essential roles in fluid homeostasis, but in the hypodermis. egl-15/FGFR mutant lethality can be rescued by constitutive activation of LET-60/Ras in the hypodermis (DeVore et al., 1995; Bulow et al., 2004). The fact that mosaic analysis of let-60 (Yochem et al., 1997) did not identify a discrete cellular focus for this lethal phenotype is consistent with a requirement for signaling in hyp7, a syncitium that derives from multiple cell lineages (Sulston et al., 1983 ). The physiological basis for egl-15/FGFR lethality or for the related “scrawny” phenotype has not been determined, but it has been speculated to involve desiccation and thus be the converse of the Clr phenotype described below (Borland et al., 2001).
Hyperactivation of egl-15/FGFR or let-60/Ras, as seen in clr-1 mutants, can result in fluid accumulation and lethality with a “Clear” (Clr) appearance, and these defects are suppressed by loss of mpk-1 or other downstream genes (the Suppressor of Clr or “Soc” phenotype) (Kokel et al., 1998; Schutzman et al., 2001). Expression studies, tissue-specific transgenes, and mosaic experiments suggest that the Clr phenotype is caused by hypodermal defects rather than excretory system defects (Huang and Stern, 2004). Indeed, the hypodermis is one major site of osmosensitive gene regulation (Lamitina et al., 2006). EGL-15 apparently plays a physiological role in osmoregulation and not a developmental role, since hyperactivation of EGL-15 at any larval stage leads to the Clr phenotype.
Germline development: mpk-1/ERK has genetically separable roles in multiple aspects of germline development, as outlined below. mpk-1 acts cell autonomously in the germline for these functions, and the pattern of MPK-1 phosphorylation in the germline is dynamic, consistent with these multiple roles (Lee et al., 2007). The entire Ras-ERK cascade is involved in many cases, as indicated, but in other cases the evidence is incomplete. Note that the relevant receptors that promote LET-60/Ras activation in the germline are unknown, although requirements for an adaptor protein, ROG-1, suggest that RTK(s) are involved at least in part (Matsubara et al., 2007) (Figure 1D).
meiotic pachytene progression: Ras-ERK signaling promotes progression through the pachytene stage of meiosis I prophase (Church et al., 1995). Null mutants have germ cells arrested at mid-pachytene (Lee et al., 2007) (Figure 3B).
pachytene cellular organization: Ras-ERK signaling promotes proper germ cell membrane organization (Lee et al., 2007; Lin and Reinke, 2008; Arur et al., 2011). Null mutants have arrested meiotic pachytene nuclei that are clumped together and lack the normal honeycomb pattern of membrane organization (Figure 3B).
germ cell sexual fate: Raf-ERK signaling promotes the male germ cell fate (Lee et al., 2007). lin-45/Raf, mek-2/MEK and mpk-1/ERK null mutants lack sperm and have a feminized germline in which all germ cells adopt a female or oocyte fate. However, these mutants fail to form normal oocytes due to the pachytene arrest and membrane disorganization phenotypes described above. Note that let-60/Ras and ksr-2 mutants do make sperm (Ohmachi et al., 2002; Lee et al., 2007). This suggests a potential Ras-independent role of Raf-MEK-ERK, although maternal rescue cannot be ruled out.
oocyte growth and differentiation: ERK signaling promotes oocyte differentiation and restrains oocyte growth (Lee et al., 2007). Mutants with reduced ERK signaling have oocytes with abnormal positions, shapes and sizes and/or oocytes that have multiple or abnormally positioned nuclei.
oocyte maturation: ERK signaling promotes sperm-dependent oocyte maturation (i.e., the transition from diakinesis to metaphase of meiosis I) (McCarter et al., 1999; Han et al., 2010; Kim et al., 2012). Maturation occurs just prior to ovulation (movement of oocytes into the spermatheca for fertilization). Mutants with reduced ERK signaling show a failure in maturation and ovulation or a significant delay in maturation, with ovulation following at the normal time (Lee et al., 2007). Note that while LIN-3-LET-23 do not function in maturation, they act by a distinct mechanism to promote ovulation (see below).
germline apoptosis: Ras-ERK signaling appears to affect germline apoptosis in multiple ways. Null mutants do not undergo physiological germline apoptosis (Gumienny et al., 1999; Kritikou et al., 2006), but show meiotic arrest prior to the late pachytene stage, when apoptosis normally occurs (Lee et al., 2007). Increasing or partially reducing MPK-1 suggests roles both in promoting (Gumienny et al., 1999; Kritikou et al., 2006) and inhibiting (Arur et al., 2009) physiological apoptosis. MPK-1 also promotes DNA-damage induced apoptosis (Rutkowski et al., 2011).
mitosis to meiosis transition: ERK signaling also appears to affect the transition from mitosis to meiosis in multiple ways. RNAi against mpk-1 leads to an expanded germline mitotic zone and a decreased early meiotic transition zone, suggesting that signaling promotes meiotic entry (Lee et al., 2006; Lee et al., 2007). On the other hand, mpk-1 null mutants have a slightly decreased mitotic zone and enhance glp-1/Notch defects (Lee et al., 2007). Note that phosphorylated MPK-1 has not been detected in the distal germline, so some of these effects could be indirect (Lee et al., 2007).
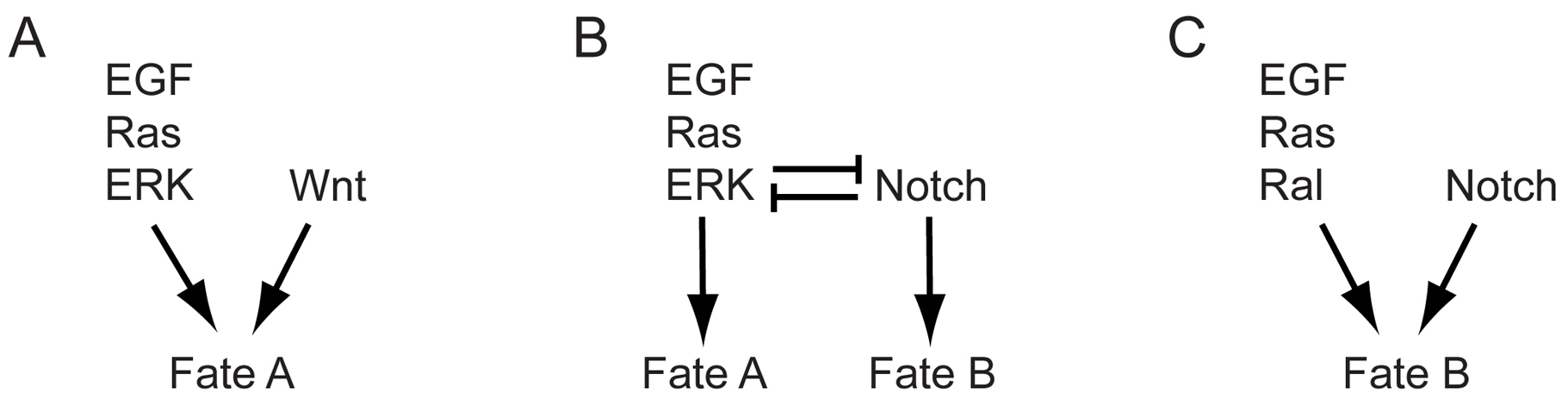
Vulval cell fates: LIN-3-LET-23 signaling via Ras cooperates with LIN-12/Notch and Wnt signaling to promote hermaphrodite vulval development (see Vulval development). Ras acts via the Raf-MEK-ERK cascade to promote the 1° vulval fate and acts via the RalGDS pathway as well as non-autonomously via Notch signaling to promote the 2° vulval fate (Simske and Kim, 1995; Chen and Greenwald, 2004; Zand et al., 2011). Reduced Ras signaling causes a vulvaless or Vul phenotype (Figure 3B), whereas increased Ras signaling leads to a multivulva or Muv phenotype. Vulvaless animals cannot lay eggs, leading to a “bag of worms” phenotype in which progeny hatch internally.
Uterine uv1 cell fate: LIN-3-LET-23 signaling via Ras-ERK promotes the uterine uv1 vs. utse cell fate (Chang et al., 1999). uv1 is important for establishing a proper vulval-uterine connection, and in its absence, hermaphrodites cannot lay eggs.
P12 ectoblast cell fate: LIN-3-LET-23 signaling via Ras-ERK cooperates with Wnt signaling to promote the P12 vs. P11 ectoblast cell fate (Fixsen et al., 1985; Jiang and Sternberg, 1998). The P11 and P12 ectoblasts are neighboring cells that give rise to slightly different types of hypodermal and neuronal descendants (Sulston and Horvitz, 1977). Reduced Ras signaling causes P12 → P11 cell fate transformations, while increased signaling causes P11 → P12 fate transformations.
Male spicule fates: LIN-3-LET-23 signaling via Ras-ERK promotes several different male spicule fates that derive from the B ectoblast (Chamberlin and Sternberg, 1994; Seah and Sternberg, 2009). B.a gives rise to four anterior-posterior pairs of cells that form equivalence groups (Sulston et al., 1980). Reduced Ras signaling causes anterior-to-posterior fate transformations within each of these equivalence groups (Chamberlin and Sternberg, 1994). The resulting spicule defects prevent males from mating effectively.
Male hook competence group fates: LIN-3-LET-23 signaling via Ras-ERK cooperates with Wnt signaling to promote the P11.p fate in the male hook competence group (HCG) (Yu et al., 2009). The HCG consists of P9.p (3° fate), P10.p (2° fate) and P11.p (1° fate) (Sulston and White, 1980). Reduced Ras signaling does not appear to affect HCG fates on its own, but can enhance the P11.p fate defects of lin-17/Frizzled mutants (Yu et al., 2009).
Aging: Ras signaling promotes longevity, potentially via multiple mechanisms. First, as animals age, LIN-3-LET-23 signaling acts via Ras-ERK and EOR-1 to upregulate skr-5 and other genes involved in the ubiquitin proteasome system (UPS), thereby promoting proper protein homeostasis (Liu et al., 2011). This pathway appears to operate broadly in epithelial cells such as the intestine and hypodermis. Second, the Raf-MEK-ERK cascade stimulates the activity of the SKN-1 transcription factor to repress insulin-like peptide expression (Okuyama et al., 2010). Whether Ras acts upstream in this process is unknown. Third, EGL-17 and EGL-15A also promote longevity, but whether they do so through Ras is unknown (Chateau et al., 2010). Loss of egl-17/FGF or egl-15A/FGFR function or RNAi knockdown of lin-45/Raf, mek-2/MEK or mpk-1/ERK shortens adult lifespan, while RNAi of the MPK-1 phosphatase gene lip-1 increases lifespan (Chateau et al., 2010; Okuyama et al., 2010). LET-23 also plays Ras-independent roles in aging (Iwasa et al., 2010) (see below).
Sex myoblast migration: EGL-17-EGL-15 signaling via Ras-ERK in sex myoblasts helps to specify the proper endpoint of sex myoblast migration (DeVore et al., 1995; Sundaram et al., 1996; Burdine et al., 1997). Two different splice isoforms of EGL-15 mediate chemoattractive and chemorepulsive mechanisms that position the SMs with respect to the gonad and vulva (Stern and Horvitz, 1991; Branda and Stern, 2000; Lo et al., 2008). In egl-15 or let-60/Ras null mosaics, sex myoblasts adopt a broadened range of final positions, whereas in egl-15A-specific mutants (which perturb chemoattraction), sex myoblasts are displaced more posteriorly (Sundaram et al., 1996; Lo et al., 2008). The resulting defects in sex muscle position can affect egg-laying.
Sex muscle differentiation: The sex myoblasts divide to generate the sex muscles, which attach to the vulva and control egg-laying (Sulston and Horvitz, 1977). Although reduced EGL-15-Ras signaling does not perturb sex muscle differentiation, hyperactive signaling by EGL-15A prevents terminal differentiation and thus causes a strong egg-laying defect (Sasson and Stern, 2004).
Body muscle maintenance: In addition to causing the Clr phenotype, hyperactivating EGL-15-Ras-ERK in adults induces body muscle wasting and failure to express a muscle gene reporter (Szewczyk et al., 2002; Szewczyk and Jacobson, 2003). This has been proposed to be a direct effect in muscle.
Axon outgrowth: LET-756-EGL-15 signaling via Ras in the hypodermis influences axon extension and pathfinding by certain ventral cord neurons that migrate over the hypodermis (Bulow et al., 2004). Whereas in wild-type these neurons extend axons along one side of the ventral midline, in mutants the axons are shorter than normal and/or wander across the midline. Whether Ras acts through Raf-MEK-ERK or through another pathway has not been determined.
Body muscle membrane extension: LET-756-EGL-15 signaling via Ras in body muscles restrains the outgrowth of muscle membrane extensions at neuro-muscular junctions (NMJs) (Dixon et al., 2006). Reduced signaling leads to ectopic muscle extensions, while increased signaling leads to fewer muscle extensions. Whether Ras acts through Raf-MEK-ERK or through another pathway has not been determined.
Acetylcholine receptor (AchR) synaptic clustering: EGL-15-Ras signaling in body muscles promotes AchR clustering and/or maintenance at NMJs (Gottschalk et al., 2005). Reduced signaling leads to reduced AchR expression at NMJs and confers moderate resistance to AchR agonists such as nicotine and levamisole. The ligand involved, and whether Ras acts through Raf-MEK-ERK or through another pathway, has not been determined.
Olfaction: Ras-ERK signaling in the mature AWC neuron is required for sensitivity to volatile attractants (Hirotsu et al., 2000). Whereas wild-type C. elegans will chemotax toward volatile attractants such as isoamylalcohol and diacetyl, mutants with reduced Ras signaling fail to chemotax towards such attractants. The receptor mediating this response is unknown, but it appears to be a G-protein coupled receptor that acts through EGL-30/Gαq to stimulate EGL-8/PLCβ and DAG production, activating the Ras GEF RGEF-1 (Chen et al., 2011) (Figure 1F).
As mentioned above, there are several contexts in which LET-23/EGFR acts via alternative Ras-independent signaling pathways (Figure 2). There also appears to be a Ras-independent mechanism for activating Raf-MEK-ERK.
Ovulation: LIN-3 and LET-23 signal through the PLCγ-IP3 pathway to promote ovulation (Clandinin et al., 1998; McCarter, 1998; Bui and Sternberg, 2002; Miller et al., 2003) (Figure 2A). LIN-3 is required in the oocyte while LET-23 is required in the somatic gonadal sheath and spermathecal cells for dilation of the distal spermatheca and contraction of the myoepithelial sheath cells necessary for ovulation. When lin-3 or let-23 activity is reduced or absent, oocytes mature but somatic gonadal function is abnormal, resulting in failed ovulation into the spermatheca, with the unfertilized oocytes undergoing endoreduplication (the Emo phenotype).
Healthy aging: LET-23 signaling through the PLCγ-IP3 pathway also promotes healthy aging (Iwasa et al., 2010). As animals age, their locomotion and swimming rates decline; this age-related decline is delayed in animals with increased LET-23 signaling and accelerated in animals with decreased signaling. The site of action of this signaling event is still unclear.
Behavioral quiescence: Over-expression of lin-3/EGF induces behavioral quiescence, a reversible, sleep-like state in which feeding and movement cease (Van Buskirk and Sternberg, 2007). This involves another Ras-independent pathway consisting of LET-23, PLC-3/PLCγ and diacylglycerol, which stimulates synaptic vesicle release in the ALA neuron (Figure 2B). let-23/EGFR and plc-3/PLCγ mutants show reduced behavioral quiescence during lethargus.
Axon maintenance: EGL-15A has a kinase- and presumably Ras-independent function in maintaining axon positions after outgrowth (Bulow et al., 2004). This function was revealed by the ability of truncated EGL-15 transgenes to rescue axon positioning defects of egl-15 mutants.
Resistance to Microbacterium nematophilum-induced swelling: ksr-1, lin-45/Raf, mek-2/MEK and mpk-1/ERK are required for the rectal swelling response to infection by M. nematophilum (Nicholas and Hodgkin, 2004). let-60/Ras is only partly required for this response, and appears to cooperate with rho-1 to activate the Raf-MEK-ERK cascade (McMullan et al, 2012).
A major strength of C. elegans as a model system is the ease of conducting unbiased forward genetic screens to identify mutants and genes relevant to a biological process of interest. Many studies on RTK-Ras-ERK signaling have focused on the role of the LET-23/EGFR pathway in promoting vulval development (see Vulval development), and many core components of the pathway were first identified through forward genetic screens for mutants with Vul or Muv mutant phenotypes (Ferguson and Horvitz, 1985). Some components of the pathway were originally identified based on other mutant phenotypes described above such as sex myoblast migration defects (Stern and Horvitz, 1991) or germline meiosis defects (Church et al., 1995; Hsu et al., 2002; Ohmachi et al., 2002). Many relevant regulators and targets of the signaling pathway were identified as genetic modifiers (suppressors or enhancers) of other mutant phenotypes in forward genetic screens or reverse genetic tests. The genes identified in these types of genetic screens have revealed many mechanisms that influence the efficiency and spatial dynamics of RTK-Ras-ERK signaling, or that mediate its downstream responses, as discussed in the following sections.
Note that for many genes described below, single mutants have relatively mild phenotypes, yet can confer strong phenotypes in certain sensitized genetic backgrounds (Table 1). Given the many detrimental effects of misregulating Ras signaling, it is perhaps not surprising that, for most steps of the signaling pathway, there are multiple layers of regulation that function semi-redundantly.
Table 2. Known MPK-1/ERK substrates. Positive targets are shown in green, and negative targets are shown in red.
| Gene Product | Mammalian Relative(s) | Molecular Function | Ras-related Phenotypes | Reference(s) |
| Soma | ||||
| EOR-1 (Egl-1 suppressor/diO uptake defective/Raf enhancer) | PLZF | BTB/Zinc finger protein, probable transcriptional regulator | Partial rod-like lethal and P12->P11 fate changes. Strong rod-like lethal in combination with sur-2, lin-25 or lin-1. Reduced age-dependent ubiquitation. | (Howard and Sundaram, 2002; Howell et al., 2010; Liu et al., 2011; Rocheleau et al., 2002) |
| LIN-1 (abnormal cell LINeage) | Elk1 | Ets domain transcription factor | Muv, excretory duct duplication, etc. Rod-like lethal in combination with eor-1 or eor-2. | (Beitel et al., 1995; Jacobs et al., 1998; Howard and Sundaram, 2002) |
| LIN-31 (abnormal cell LINeage) | FoxB2 | Winged helix transcription factor | Mixed Vul and Muv | (Miller et al., 1993; Tan et al., 1998) |
| Germline | ||||
| CDC-48.2 (Cell Division Cycle related) | p97 | AAA ATPase | Enhances mpk-1(rf) pachytene organization and oocyte differentiation defects | (Arur et al., 2009) |
| CDK-7 (Cyclin-Dependent Kinase) | CDK7/CAK | Kinase | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| CDTL-7 CDc-Two-Like) | CRKRS/CDTL7 | Kinase | Enhances mpk-1(rf) pachytene progression and apoptosis defects | (Arur et al., 2009) |
| CGH-1 (Conserved Germline Helicase) | DDX6 | ATP-dependent RNA Helicase | Enhances mpk-1(rf) pachytene progression and apoptosis defects | (Arur et al., 2009) |
| CYA-1 (CYclinA) | CYA1 | Cyclin A | Enhances mpk-1(rf) oocyte differentiation defects | (Arur et al., 2009) |
| DCR-1 (DiCeR) | DICER | small RNA synthesis | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| DIS-3 (DISjunction abnormal) | DIS3 | exosome catalytic subunit | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| DPL-1 (DP-Like) | DP | component of E2F transcription factor | Suppresses mpk-1(lf) pachytene disorganization defect | (Lin and Reinke, 2008) |
| DRSH-1 (DRoSHa) | DROSHA | small RNA synthesis | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| DDX-19 (DeaD boX helicase) | DDX19 | Dead box helicase | Enhances mpk-1(rf) pachytene organization and oocyte differentiation defects | (Arur et al., 2009) |
| EIF-3.D (Eukaryotic Initiation Factor) | EIF3 | Translation initiation Factor | Enhances let-60(gf) oocyte maturation defect | (Arur et al., 2009) |
| GSK-3 (Glycogen Synthase Kinase) | GSK3B | Serine/Threonine Kinase | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| MRPS-5 (Mitochondrial Ribosomal Protein, Small) | MRPS5 | Ribosomal Protein S5 | Enhances mpk-1(rf) pachytene progression and oocyte differentiation defects | (Arur et al., 2009) |
| MTR-4 (MRNA Transporter) | SKIV-2L2 | Nuclear exosomal RNA helicase | Enhances mpk-1(rf) pachytene progression and pachytene organization defects | (Arur et al., 2009) |
| NOS-3 (NanOS-related) | - | RNA binding protein | Suppresses mpk-1(lf) pachytene disorganization defect | (Arur et al., 2011) |
| PAC-1 (PAR-6 At Contacts) | ARHGAP21 | Rho GAP | Enhances mpk-1(rf) pachytene progression and oocyte differentiation defects | (Arur et al., 2009) |
| PAR-5 (abnormal embryonic PARtitioning of cytoplasm) | 14-3-3-zeta | 14-3-3 family chaperone | Enhances mpk-1(rf) pachytene progression and pachytene organization defects | (Arur et al., 2009) |
| POLE-2 (DNA POLymerase Epsilon) | POLE2 | DNA Polymerase epsilon, subunit B | Enhances mpk-1(rf) oocyte growth defects | (Arur et al., 2009) |
| RAB-5 (RAB family) | RAB5 | Rab GTPase | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| RBA-1 (RBAp48-related) | CAF1/TBAP48 | Rb-binding protein | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| RHA-2 (RNA HelicAse) | DHX37 | RNA helicase | Enhances mpk-1(rf) pachytene progression and oocyte growth defects | (Arur et al., 2009) |
| ROP-1 (RO ribonucleoProtein) | R(O) RNP | RNA binding protein | Enhances let-60(gf) small oocyte defect | (Arur et al., 2009) |
| RSKD-1 (Ribosomal protein S6 Kinase Delta) | RPS6K | Ribosomal Protein S6 Kinase | Enhances mpk-1(rf) maturation defects | (Arur et al., 2009) |
| RSKN-1 (RSK-pNinety) | RSK4 | Ribosomal Protein S6 Kinase | Enhances mpk-1(rf) pachytene progression and oocyte differentiation defects | (Arur et al., 2009) |
| SEL-11/HRD-1 (Suppressor/Enhancer of LIN-12) | SYVN1 | E3 ubiquitin ligase | Enhances mpk-1(rf) pachytene organization and oocyte differentiation defects | (Arur et al., 2009) |
| TOE-1 (Target of ERK kinase MPK-1) | HEAT1 | HEAT-repeat protein | Enhances mpk-1(rf) pachytene organization and oocyte defects | (Arur et al., 2009) |
| TOE-2 (Target of ERK kinase MPK-1) | DEPDC1B | DEP-domain protein | Enhances mpk-1(rf) apoptosis defects | (Arur et al., 2009) |
| TOE-4 (Target of ERK kinase MPK-1) | RNF44 | RING finger protein, predicted E3 ubiquitin ligase | Enhances mpk-1(rf) oocyte differentiation defect | (Arur et al., 2009) |
| TOE-5 (Target of ERK kinase MPK-1) | HMMR | F-box protein | Enhances mpk-1(rf) apoptosis defects | (Arur et al., 2009) |
| TTBK-2 (Tau TuBulin Kinase) | TTBK2 | Casein kinase | Enhances mpk-1(rf) apoptosis defects | (Arur et al., 2009) |
| USP-46 (Ubiquitin Specific Protease) | USP46 | ubiquitin protease | Enhances mpk-1(rf) oocyte differentiation defect | (Arur et al., 2009) |
| ZIM-2 (Zinc Finger in Meiosis) | ZFP62 | C2H2 zinc finger protein | Enhances mpk-1(rf) germline sex determination defects | (Arur et al., 2009) |
Transcriptional regulation of growth factor ligand expression is an important determinant of when and where RTK signaling occurs in C. elegans (Figure 4). lin-3/EGF, egl-17/FGF and let-756/FGF all show relatively restricted patterns of gene expression. For example, lin-3 is expressed in the gonadal anchor cell during vulval development (Hill and Sternberg, 1992), in primary vulval descendants during uterine development (Chang and Sternberg, 1999), and in the excretory canal cell during excretory duct development (Abdus-Saboor et al., 2011). egl-17 is expressed in the gonad and in the primary vulval precursor cell P6.p during sex myoblast migration (Burdine et al., 1998; Branda and Stern, 2000). let-756 is expressed in body muscles and some neurons (Bulow et al., 2004). egl-17 expression in P6.p and lin-3 expression in P6.p descendants both depend on LET-23/EGFR and LET-60/Ras-dependent cell fate specification (Burdine et al., 1998; Chang and Sternberg, 1999; Cui and Han, 2003). Simple overexpression of lin-3(+) is sufficient to cause gain-of-function phenotypes (Katz et al., 1995), and multiple genes that restrict or promote lin-3 expression have been identified.
Syn Muv genes: The “synthetic multivulva” (SynMuv) genes encode three sets of nuclear factors that function redundantly to inhibit ectopic lin-3 expression (Ferguson and Horvitz, 1989; Cui et al., 2006; Fay and Yochem, 2007). One set, the SynMuv B genes, encodes orthologs of a conserved transcriptional repression cascade including LIN-35/Rb (Lu and Horvitz 1998), DPL-1/DP and EFL-1/E2F (Ceol and Horvitz, 2001), a NuRD-like histone deacetylase complex including HDA-1/HDAC1 (Solari and Ahringer, 2000), an H3K9 histone methyltransferase MET-2/SETDB1 (Andersen and Horvitz, 2007) and the chromodomain protein HPL-2/HP1 (Couteau et al., 2002). The SynMuv C genes encode components of a Tip60/NuA4-like histone acetyltransferase complex (Ceol and Horvitz, 2004), while the SynMuv A genes include several THAP-domain proteins that are thought to act as transcriptional repressors (Huang et al., 1994; Davison et al., 2005; Davison et al., 2011). A point mutation in the lin-3 gene promoter behaves like a SynMuv A mutation, suggesting that synMuv A proteins may act directly on the lin-3 promoter to repress ectopic lin-3 expression (Saffer et al., 2011). It is not clear if the other SynMuv complexes act directly or indirectly on lin-3. Although single mutants for any given SynMuv gene have relatively normal levels of lin-3 expression, double mutants for a SynMuv A and a SynMuv B mutation have increased levels of lin-3 expression and several corresponding Ras(gf)-like phenotypes such as Muv (Cui et al., 2006). Single molecule RNA FISH demonstrated that SynMuv mutants have ectopic lin-3 mRNA in most cells of the body, but that elevation in lin-3 mRNA levels is quite modest on a per cell basis, highlighting the sensitivity of signaling to ligand levels (Saffer et al., 2011). Functional analysis suggests that the Muv phenotype primarily results from derepression of lin-3 in hyp7 (Myers and Greenwald, 2005; Cui et al., 2006).
Transcription factors that promote lin-3 expression: In the anchor cell, lin-3 expression requires the E-protein/daughterless homolog HLH-2 and an unidentified nuclear hormone receptor (NHR); a 59 bp lin-3 enhancer element containing predicted E-box and NHR binding sites is both necessary and sufficient for anchor cell expression (Hwang and Sternberg, 2004). In the primary vulval lineage, lin-3 expression requires the Pax family transcription factor EGL-38 (Chang and Sternberg, 1999).
Proteolytic processing and secretion of ligands and interaction of ligands with receptors are additional steps at which signaling can be regulated (Figure 4).
LIN-3/EGF proteolysis and signaling: Like other ligands of the EGF family, LIN-3 is synthesized initially as a transmembrane protein but may be subsequently cleaved proteolytically to generate a diffusible form (Hill and Sternberg, 1992; Blobel et al., 2009). There are at least four different splice isoforms of LIN-3 that differ in their juxta-membrane regions, potentially affecting proteolysis (Dutt et al., 2004; Van Buskirk and Sternberg, 2007). Biological activity of one specific isoform, LIN-3L, appears to require proteolysis by the Rhomboid ortholog ROM-1 (Dutt et al., 2004). rom-1 mutants have only subtle phenotypes, suggesting either that other LIN-3 proteases exist or that other LIN-3 isoforms can function effectively in an unprocessed, transmembrane form.
In Drosophila, various extracellular factors influence EGF-like ligand diffusion and interactions with receptors (Klein et al., 2004; Miura et al., 2006; Pizette et al., 2009). There is emerging evidence for this type of regulation in C. elegans as well. The secreted proteins HPA-1 and HPA-2 limit LIN-3 signaling during adult aging, potentially by LIN-3 sequestration (Iwasa et al., 2010). The secreted lipocalin LPR-1 facilitates LIN-3 signaling during excretory duct cell development (C. Stone and M. Sundaram, in preparation).
FGF secretion and signaling: Both C. elegans FGF-like ligands utilize unusual pathways for secretion. EGL-17 is secreted via a mechanism that requires the lipoprotein receptor-related proteins LRP-1 and LRP-2 and the Disabled-related adaptor DAB-1 (Kamikura and Cooper, 2003). LET-756 lacks a classical signal peptide, and like mammalian FGF9, its secretion depends on an internal hydrophobic domain (Popovici et al., 2004). Also like FGF9, LET-756 shows partial localization to the nucleus (Popovici et al., 2006), though the significance of this localization remains unknown.
In C. elegans, as in other organisms, FGF signaling can be influenced by the extracellular matrix (Bulow and Hobert, 2006). Heparan sulfate polysaccharides facilitate EGL-17-dependent axon outgrowth (Tornberg et al., 2011). The matrix component unc-52/perlecan genetically interacts with egl-17 to influence distal tip cell migration (Merz et al., 2003). The ability of EGL-17 vs. LET-756 to signal through EGL-15/FGFR also depends to some degree on alternative splicing of egl-15 (see below).
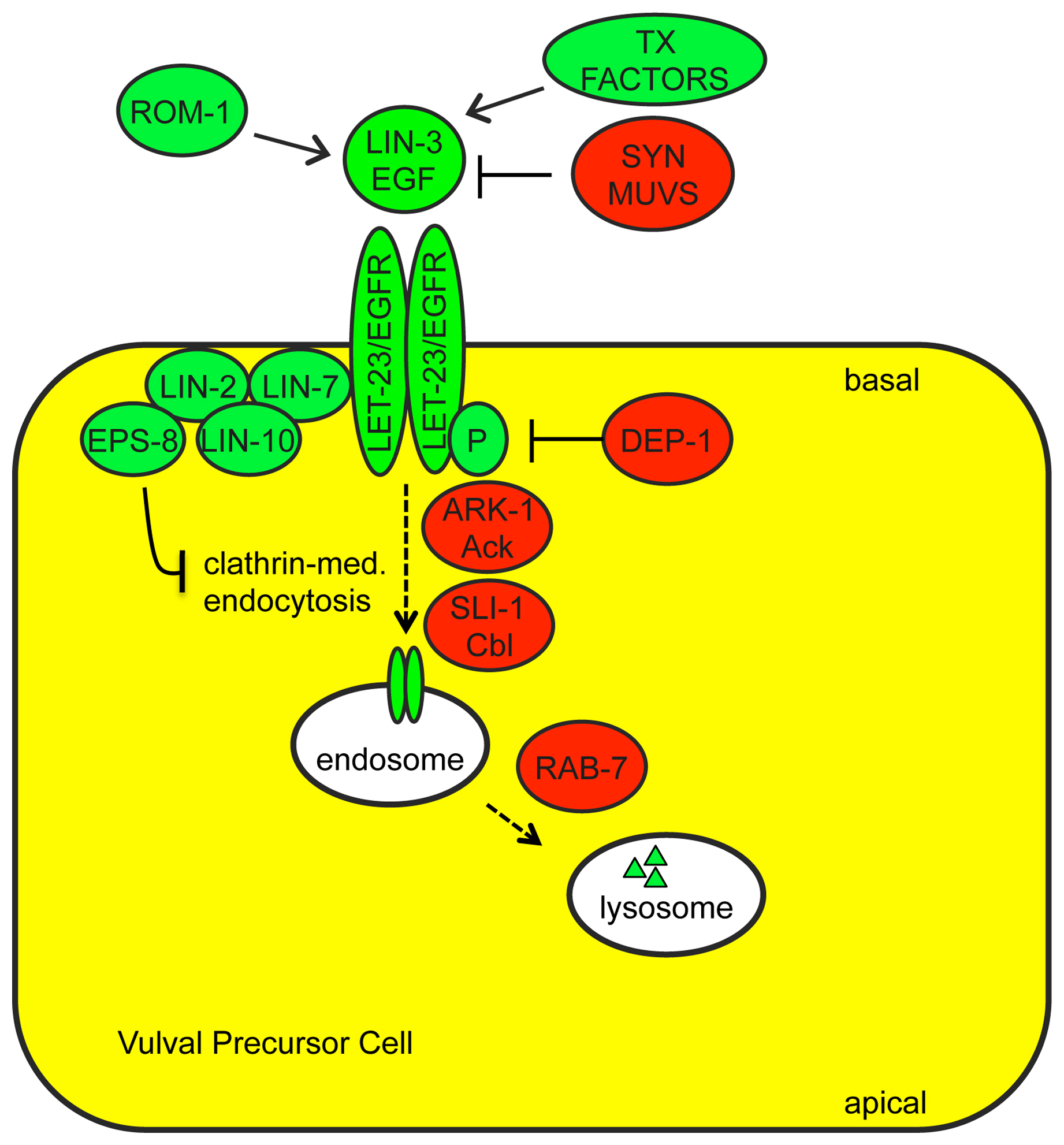
LET-23/EGFR basolateral localization: Factors that regulate LET-23/EGFR trafficking and localization are important modulators of signaling strength during vulva development (Figure 4). A complex consisting of three PDZ-domain proteins, LIN-2, LIN-7 and LIN-10, facilitates signaling by localizing LET-23 to the basolateral membrane of vulval precursor cells, adjacent to the source of LIN-3 ligand (Simske et al., 1996; Kaech et al., 1998; Whitfield et al., 1999). EPS-8 (EGFR substrate protein −8) interacts with this LIN-2-LIN-7-LIN-10 complex to further enhance basolateral LET-23 levels in the primary vulval precursor cell (Stetak et al., 2006). The E3 ubiquitin ligase SLI-1/Cbl (Jongeward et al., 1995; Yoon et al., 1995), the Ack-related tyrosine kinase ARK-1 (Hopper et al., 2000), the clathrin adaptors UNC-101 (Lee et al., 1994) and DPY-23 (Yoo et al., 2004), the sorting nexin LST-4 (Yoo et al., 2004), the RAB-7 GTPase and several members of the ESCRT complex (Skorobogata and Rocheleau, 2012) all negatively regulate signaling, probably by promoting LET-23 endocytosis and lysosomal degradation.
EGL-15/FGFR alternative splicing and glycosylation: EGL-15/FGFR activity is regulated in part via alternative splicing and other modifications to its extracellular domain that may affect ligand binding. The EGL-15(A) and EGL-15(B) splice isoforms differ in a C. elegans-specific region of the extracellular domain, and EGL-15(A) primarily mediates EGL-17/FGF-dependent signaling, while EGL-15(B) primarily mediates LET-756/FGF-dependent signaling (Goodman et al., 2003; Lo et al., 2008). However, these functional distinctions are not complete, and both ligands can act to some degree through both receptor isoforms (Goodman et al., 2003; Bulow et al., 2004; Dixon et al., 2006). At least five other alternative splice forms differ in the EGL-15 extreme C-terminus, potentially affecting interactions with downstream targets. EGL-15 is N-glycosylated in vivo and this modification appears to negatively regulate its activity, potentially by interfering with ligand binding or self-association (Polanska et al., 2009). EGL-15A activity may be positively regulated by two C. elegans orthlogs of Klotho, a mammalian FGFR binding protein and co-receptor (Chateau et al., 2010; Polanska et al., 2011).
Adaptor Proteins that bind RTKs: RTKs transduce signals by autophosphorylation and subsequent binding to cytosolic phospho-tyrosine binding adaptor proteins, which then recruit other factors (Lemmon and Schlessinger, 2010). LET-23 acts primarily through the adaptor SEM-5 (related to vertebrate Grb2) (Clark et al., 1992), which recruits SOS-1 (Downward, 1994), but also in part through SOC-1 (related to mammalian Gab1), which recruits PTP-2/Shp2 (Hopper, 2006) (Figure 1B). EGL-15 acts through both SEM-5 and SOC-1-PTP-2 (Schutzman et al., 2001) (Figure 1C). Whether these RTKs interact directly or indirectly with SOC-1 (via SEM-5) has not been established. The SOC-1 adaptor also acts with the RTK SCD-2/ALK (Reiner et al., 2008). The ROG-1 adaptor (related to vertebrate FRS2) does not seem to act downstream of LET-23 or EGL-15, but may act downstream of another unknown RTK to control Ras-ERK-dependent meiotic progression in the germline (Matsubara et al., 2007; Lo et al., 2010) (Figure 1D). Adaptors can influence the relative use of different RTK-dependent signaling pathways, as SEM-5 and SOC-1 promote LET-23-mediated activation of LET-60 Ras, but inhibit LET-23-mediated activation of the PLCγ-IP3 pathway (Hopper et al., 2000; Hopper, 2006).
Receptor Tyrosine Phosphatases: Factors that regulate RTK phosphorylation status could obviously modulate adaptor binding and signaling strength. The receptor tyrosine phosphatases (RTPs) CLR-1 and DEP-1 negatively regulate EGL-15 and LET-23 RTK activity, respectively, most likely through direct dephosphorylation of key tyrosine residues (Kokel et al., 1998; Berset et al., 2005). CLR-1 is related to the LAR RTP family, and is required in the hypodermis to prevent EGL-15/FGFR hyperactivation (Kokel et al., 1998; Huang and Stern, 2004) (Figure 1C). DEP-1 is the C. elegans ortholog of the mammalian tumor suppressor Density Enhanced Phosphatase-1, which inhibits EGFR and multiple other RTKs (Ruivenkamp et al., 2002; Tarcic et al., 2009). DEP-1 acts redundantly with a variety of other inhibitors to dampen LET-23/EGFR signaling (Berset et al., 2005) (Figure 4).
Cytosolic phosphatases and kinases: The cytosolic tyrosine phosphatase PTP-2/Shp2 positively regulates LET-23 and EGL-15 RTK signaling (Gutch et al., 1998; Schutzman et al., 2001), and the cytosolic tyrosine kinase ARK-1 (Ack-related) negatively regulates LET-23 RTK signaling (Hopper et al., 2000). The substrates of PTP-2 and ARK-1 are still unclear, but these proteins can bind to the adaptors SOC-1 and SEM-5, respectively, and appear to act at a step immediately downstream of the RTKs to influence the strength or kinetics of signaling. In the germline, PTP-2 antagonizes the activity of GAP-2 and GAP-3, suggesting these could be relevant PTP-2 substrates (Yang et al., 2010). Evidence from mammalian cells suggests that Ack tyrosine kinases promote EGFR internalization and degradation (Shen et al., 2007).
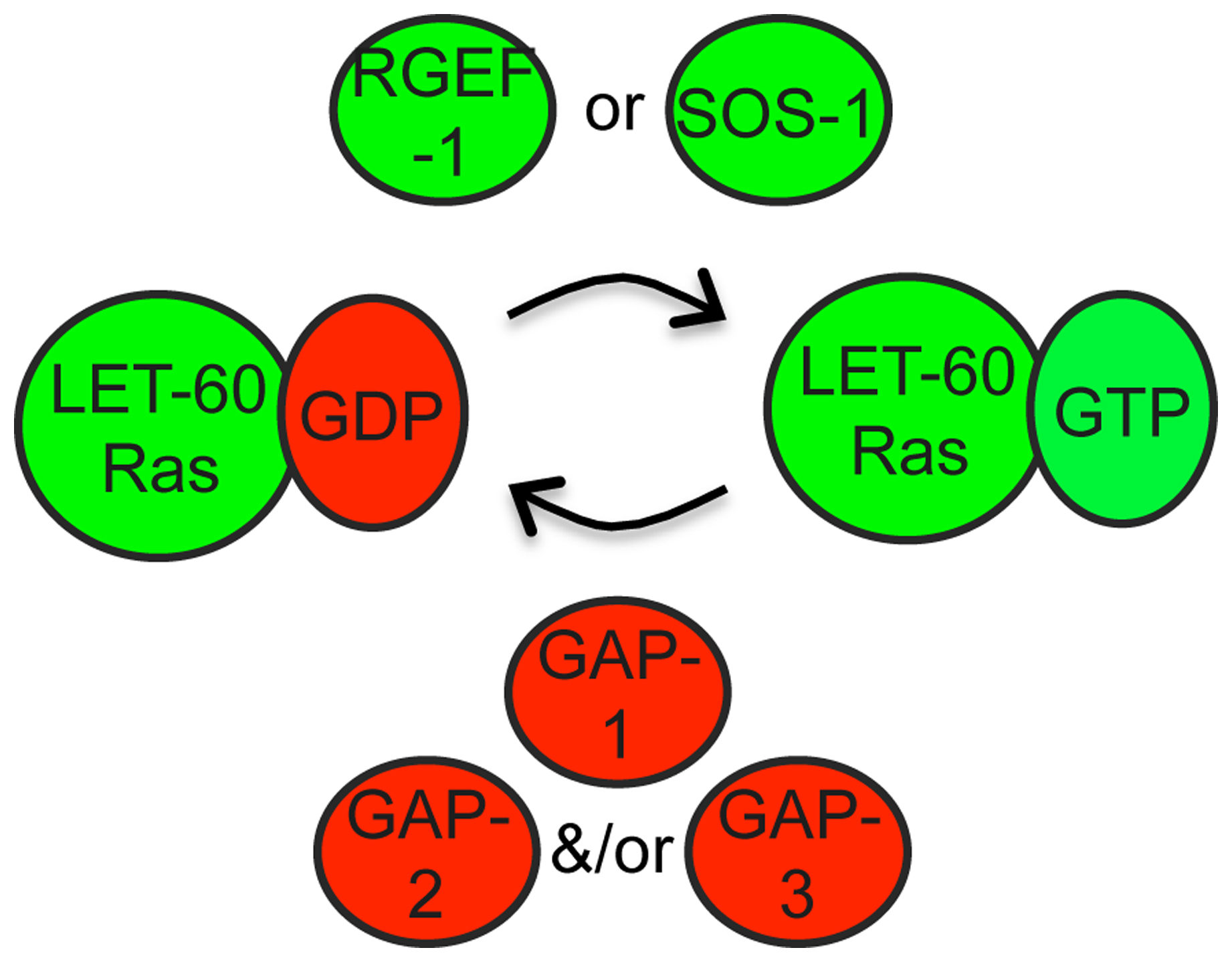
GEFs and GAPs: As is the case for other small GTPases, the activity of LET-60/Ras is controlled by Guanine Nucleotide Exchange Factors (GEFs), which activate Ras by stimulating conversion of Ras-GDP to Ras-GTP, and by GTPase activating proteins (GAPs), which inactivate Ras by stimulating conversion of Ras-GTP to Ras-GDP (Malumbres and Barbacid, 2002; Karnoub and Weinberg, 2008) (Figure 5). Gain-of-function (gf) mutations lock Ras in the active, GTP-bound state; the let-60(gf) allele n1046 (G13E) has been widely used for genetic analyses (Han and Sternberg, 1990). Dominant-negative (dn) mutations lock Ras in the inactive, GDP-bound state, causing it to bind stably to and titrate out GEFs; a variety of let-60(dn) mutations have been described (Han and Sternberg, 1991).
|
Figure 5. Regulation of LET-60/Ras by GEFs and GAPs. Ras GTPase regulation by GEFs and GAPs has been extensively studied in other systems (Malumbres and Barbacid, 2002). The GEF SOS-1 has widespread requirements in LET-60/Ras signaling (Chang et al., 2000). The alternative RGEF-1 is used in AWC (Chen et al., 2011). There are three C. elegans GAP proteins that show varying tissue specificity and function in a semi-redundant manner (Stetak et al., 2008).
The GEF SOS-1 is required for most Ras-mediated developmental events (Chang et al., 2000). A temperature-sensitive allele, sos-1(cs41), is a useful tool for conditional inactivation of Ras signaling (Rocheleau et al., 2002). A different GEF, RGEF-1 (related to mammalian RasGRPs), regulates LET-60 Ras in AWC neurons during olfaction (Chen et al., 2011).
Three GAPs, GAP-1, GAP-2 and GAP-3, function redundantly to negatively regulate Ras signaling, with each required to different degrees in different tissues. GAP-1 plays a predominant role during vulval and P12 fate specification, while GAP-3 predominates in the germline (Hajnal et al., 1997; Hayashizaki et al., 1998; Stetak et al., 2008).
Post-translational modifications of Ras: In other systems, a variety of post-translational modifications can modulate Ras trafficking and localization (Ahearn et al., 2012). For example, farnesylation of the Ras C-terminus promotes plasma membrane attachment. Chemical farnesyltransferase inhibitors suppress the Muv phenotype of let-60 ras(gf) mutants, suggesting that farnesylation promotes LET-60/Ras activity (Hara and Han, 1995). A negative regulator of LET-60/Ras is SUR-5, a protein of unknown function that resembles acetyl coenzyme A synthetases (Gu et al., 1998).
miRNA-dependent regulation of Ras: The overall level of let-60 ras expression can also influence Ras signaling activity, as overexpression of wild-type LET-60 leads to a Muv phenotype (Sundaram and Han, 1995). In vulval precursor cells and hypodermal seam cells, normal levels of let-60 ras depend on negative regulation by the let-7 family microRNAs mir-84 and let-7 (Johnson et al., 2005). These microRNAs appear to act directly on the let-60 3’ UTR, and this mode of regulation is conserved in mammalian ras genes (Johnson et al., 2005).
Raf, MEK and ERK activities depend in large part on their phosphorylation status (Udell et al., 2011). For example, Raf must be de-phosphorylated at certain sites and phosphorylated at other sites in order to become active (Chong et al., 2001) (Figure 6A). Transgenic, hyperactive forms of LIN-45/Raf, MEK-2/MEK and MPK-1/ERK were generated by mutating relevant sites to prevent or mimic phosphorylation; such transgenes have been useful for epistasis experiments (Lackner and Kim, 1998; Chong et al., 2001; Kao et al., 2004; Yoder et al., 2004; Rocheleau et al., 2005). Commercially available antibodies also recognize phosphorylated MPK-1 by Western analyses and (in some cases) immunofluorescence, and have been very useful for molecular epistasis (Hirotsu et al., 2000; Page et al., 2001; Ohmachi et al., 2002; Lee et al., 2007; Chen et al., 2011).
|
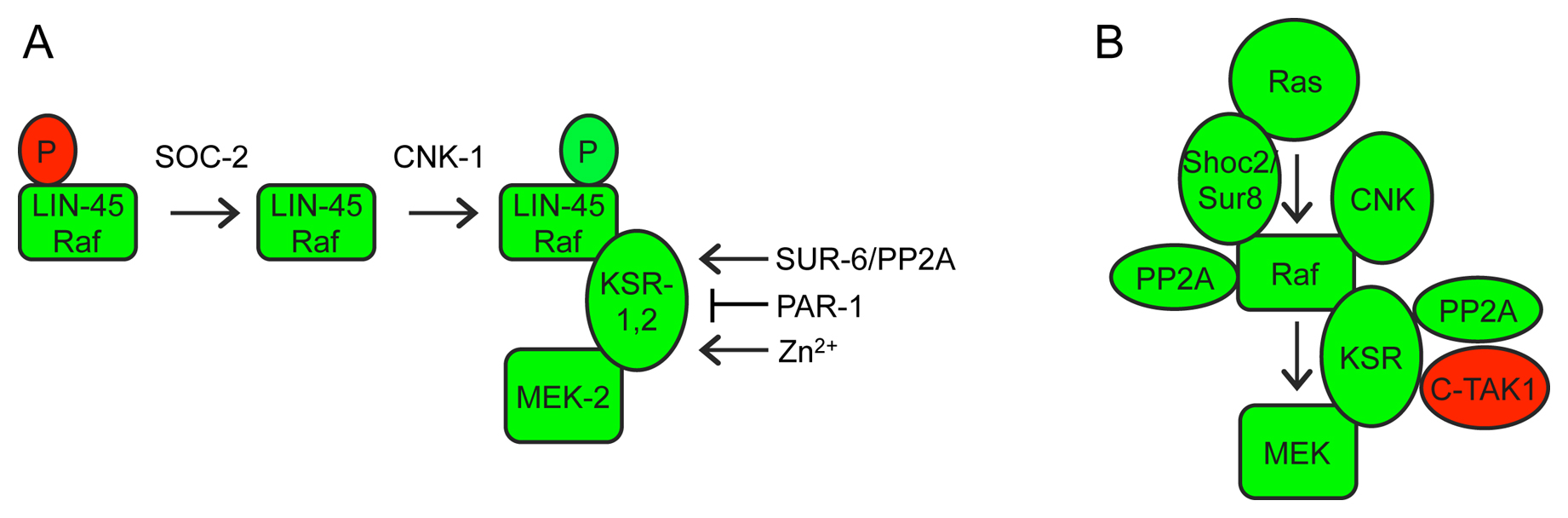
Figure 6. Regulation of Raf-MEK-ERK by scaffolds and other associated proteins. A) Phosphorylation (P) at different sites inhibits (red) or activates (green) LIN-45 and other Raf proteins (Chong et al., 2001; Udell et al., 2011). Epistasis experiments with LIN-45/Raf phosphorylation site variants demonstrated that SOC-2 is required upstream or during inhibitory phosphorylation removal (Yoder et al., 2004), while CNK-1 is required downstream of inhibitory phosphate removal but upstream of activating phosphorylation addition (Rocheleau et al., 2005). KSR-1 and SUR-6 are required downstream of both events (Kao et al., 2004; Yoder et al., 2004; Rocheleau et al., 2005). B) Protein interactions of related scaffold proteins in mammalian cells (Udell et al., 2011). C-TAK1 is a mammalian homolog of the PAR-1 kinase (Muller et al., 2001). See text for additional details and references.
KSR and other scaffold proteins: Positive regulators of Raf-MEK-ERK include so-called “scaffold” proteins (Figure 6A) - KSR-1 (Kornfeld et al., 1995b; Sundaram and Han, 1995), KSR-2 (Ohmachi et al., 2002), CNK-1 (Rocheleau et al., 2005) and SOC-2 (also known as SUR-8) (Selfors et al., 1998; Sieburth et al., 1998). These proteins have no obvious enzymatic activity, but bind to one or more core kinases in the cascade and are thought to bring different components together at the right place and time (Udell et al., 2011). KSR-1 and KSR-2 are required downstream of activating Raf phosphorylation but upstream of MEK-ERK (Ohmachi et al., 2002; Rocheleau et al., 2005). CNK-1 is required upstream of Raf activating phosphorylation (Rocheleau et al., 2005), while SOC-2 is required upstream of (or during) Raf inhibitory phosphate removal (Yoder et al., 2004). These epistasis results fit well with the observed protein interactions of the mammalian scaffolds (Figure 6B): Mammalian KSR binds constitutively to MEK and variably to Raf and ERK (Xing et al., 1997; Yu et al., 1998; Stewart et al., 1999), while CNK binds Raf (Yao et al., 2000; Lanigan et al., 2003), and SOC-2 binds to Ras and Raf (Li et al., 2000). In C. elegans, the KSR scaffold is a core pathway component but encoded by two paralogs: KSR-1 and KSR-2 are individually required for a few developmental processes, but redundantly required for many others (Ohmachi et al., 2002). CNK-1 and SOC-2 are not individually required under standard laboratory conditions, but help buffer signaling against other perturbations (Selfors et al., 1998; Sieburth et al., 1998; Rocheleau et al., 2005). Another positive regulator of Raf-MEK-ERK signaling is CGR-1, a CRAL/TRIO and GOLD domain protein that promotes signaling in the vulva, possibly by modulating Raf activation (Goldstein et al., 2006; Johnson and Kornfeld, 2010).
Regulators of the KSR scaffold: Several gene products appear to influence KSR scaffold activity (Figure 6). The PAR-1 kinase inhibits Raf-MEK-ERK signaling (Kao et al., 2004; Yoder et al., 2004) and is homologous to mammalian C-TAK1, which directly phosphorylates KSR to affect its localization (Ory et al., 2003). SUR-6, a PR55/B-type regulatory subunit of Protein Phosphatase 2A (PP2A-B), promotes Raf-MEK-ERK signaling (Sieburth et al., 1999) and has been suggested to dephosphorylate and activate both Raf and KSR (Ory et al., 2003; Kao et al., 2004; Yoder et al., 2004). Finally, the zinc transporter proteins CDF-1 and SUR-7 also promote Raf-MEK-ERK signaling in the vulva (Bruinsma et al., 2002; Yoder et al., 2004). High levels of zinc can influence KSR phosphorylation in mammalian cells, suggesting that zinc modulates the activity of a KSR kinase (Yoder et al., 2004).
MPK-1/ERK inhibitors: Several inhibitors of MPK-1/ERK have been described. The MAP kinase phosphatase LIP-1 dephosphorylates and inhibits MPK-1 in both the soma and the germline (Berset et al., 2001; Hajnal and Berset, 2002; Lee et al., 2006; Rutkowski et al., 2011). The CCCH zinc finger protein GLA-3 binds to and inhibits MPK-1 without altering MPK-1 phosphorylation (Kritikou et al., 2006). The Germinal Center Kinase GCK-1 also binds directly to MPK-1 and inhibits MPK-1 phosphorylation in the pachytene region of the germline (Schouest et al., 2009). A gap junction-dependent mechanism and a VAB-1 RTK-dependent mechanism inhibit MPK-1 phosphorylation in oocytes (Miller et al., 2003; Govindan et al., 2006; Govindan et al., 2009; Kim et al., 2012) (Figure 7). Repression of mpk-1 expression by the RNA binding proteins FBF-1, FBF-2 and LARP-1 also helps shape the distribution of phosphorylated MPK-1 in the germline (Lee et al., 2007; Nykamp et al., 2008).
|
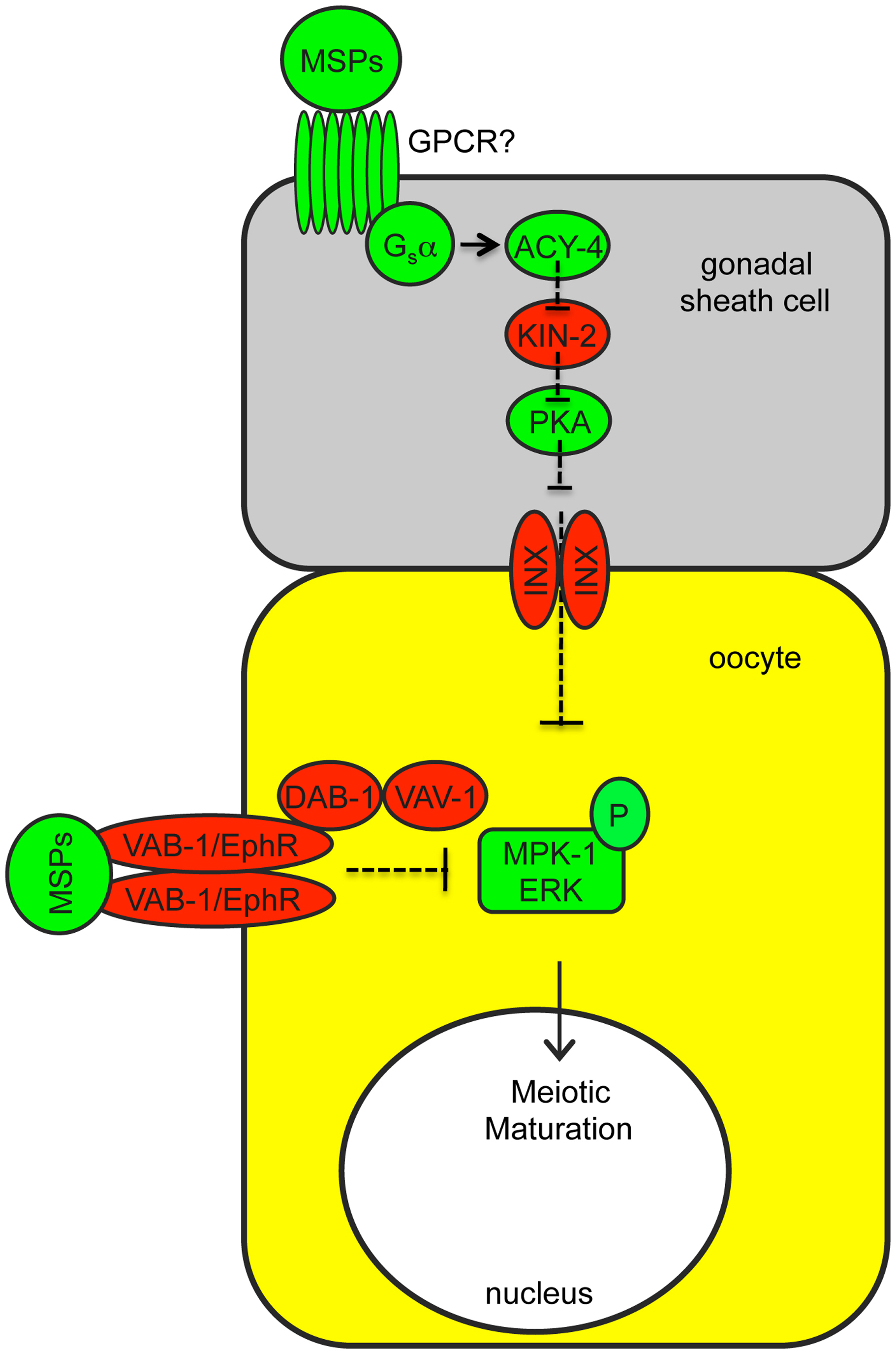
Figure 7. Regulation of MPK-1/ERK phosphorylation during oocyte maturation. Sperm, via the Major Sperm Protein (MSP), promotes MPK-1/ERK phosphorylation and oocyte maturation (McCarter et al., 1999; Miller et al., 2001; Miller et al., 2003). Somatic gonadal sheath cells signal adjacent oocytes via innexin (INX)-based gap junctions, antagonizing MPK-1/ERK activation (Govindan et al., 2006; Govindan et al., 2009; Kim et al., 2012). When sperm are present, MSP relieves gap junctional inhibition by binding to unknown G-protein coupled receptors (GPCRs) on sheath cells that act through Gsα and ACY-4 adenylate cyclase signaling and Protein Kinase A (PKA). The VAB-1 RTK in oocytes also antagonizes MPK-1/ERK activation via a mechanism that involves DAB-1/Disabled and VAV-1/Vav, and this is also relieved by MSP (Miller et al., 2003; Govindan et al., 2006). It is currently unknown what pathway directly activates MPK-1 in oocytes or at what step gap junction-dependent and VAB-1 signaling antagonize MPK-1 activation.
Cross-tissue interactions: There is evidence that some genes may influence Raf-MEK-ERK signaling indirectly, by affecting neighboring cells. Some of the clearest examples involve genes that function within gonadal sheath cells to affect gap-junction dependent inhibition of MPK-1/ERK phosphorylation in oocytes (Kim et al., 2012) (Figure 7). Also, the Gαq protein EGL-30 and the voltage-gated calcium channel EGL-19 function in neurons and muscle, respectively to promote Ras-mediated vulval induction (Moghal et al., 2003). The G-protein coupled receptor SRA-13 and its Gα target GPA-5 inhibit Ras-mediated vulval induction, but it is unknown whether these genes function in vulval cells or in neurons (Battu et al., 2003). The zinc transporter CDF-1 can influence vulval induction when expressed in either the vulva or the intestine (Bruinsma et al., 2002). The mechanisms underlying these other cross-tissue effects are still unclear.
The specific outcomes of RTK-Ras-ERK signaling in different tissues are thought to depend on the availability and use of different ERK substrates and on combinatorial action of ERK substrates with other tissue-specific or signal-regulated factors (Tan and Kim, 1999; Flores et al., 2000; Halfon et al., 2000; Barolo and Posakony, 2002; Arur et al., 2009). In C. elegans somatic cell fate decisions, the combinatorial mechanism appears quite relevant, since two identified MPK-1/ERK targets (LIN-1 and EOR-1) are widely expressed and required for many different outcomes (Beitel et al., 1995; Jacobs et al., 1998; Howard and Sundaram, 2002; Rocheleau et al., 2002; Howell et al., 2010) (Figure 1B; Table 2). LIN-1 and EOR-1 are both nuclear transcription factors, consistent with the idea that transcriptional control of gene expression is the critical mechanism for cell fate specification. The situation is different in the germline, where there are many identified targets of MPK-1, few of which are transcription factors and each of which affects only a small subset of MPK-1-dependent processes (Arur et al., 2009) (Figure 1D; Table 2). In both the soma and the germline, a common theme is redundancy among different MPK-1/ERK substrates.
LIN-1/Ets: The Ets domain transcription factor LIN-1 is arguably the most important MPK-1 substrate in the soma (Beitel et al., 1995; Jacobs et al., 1998) (Figure 1B; Table 2). LIN-1 is related to the mammalian ERK target Elk1 (Sharrocks, 2002; Yang et al., 2003). LIN-1 is a direct substrate of ERK in vitro, and studies of lin-1 gain-of-function mutations first identified the FXF ERK docking site, which confers high affinity binding between ERK and many of its substrates, including Elk1 (Jacobs et al., 1998; Jacobs et al., 1999; Sharrocks et al., 2000). lin-1 null mutants have phenotypes resembling those of let-60 ras gain-of-function mutants and are epistatic to mpk-1, suggesting that Ras-ERK signaling antagonizes LIN-1 activity (Beitel et al., 1995; Lackner and Kim, 1998). However, some evidence suggests that LIN-1 has an additional positive role in Ras signaling, since removal of lin-1 in certain genetic backgrounds can cause Ras loss-of-function phenotypes (Howard and Sundaram, 2002; Tiensuu et al., 2005; Abdus-Saboor et al., 2011) (see below). The biochemical basis for these opposite roles is not yet clear. One simple model consistent with studies of Elk1 (Boyer et al., 1999; Yang et al., 2003) is that MPK-1/ERK phosphorylation converts LIN-1 from a transcriptional repressor to a transcriptional activator in order to turn on signal-dependent gene expression (Figure 1B). Alternatively, MPK-1/ERK phosphorylation may simply inactivate LIN-1, and unphosphorylated LIN-1 may repress or activate different targets. The dual positive and negative roles of LIN-1 could indicate that LIN-1 has some targets that have positive roles and some targets that have negative roles.
There is evidence for LIN-1 having repressor activity in cell culture and in vivo (Leight et al., 2005). The repressive role of LIN-1 requires sumoylation, which then allows a SUMO-dependent interaction with MEP-1, a component of the C. elegans NuRD-like repressor complex (Leight et al., 2005). LIN-1 also physically associates with the NuRD-like component LET-418/Mi-2 (Guerry et al., 2007). Two directly repressed targets of LIN-1 in the vulva are the Hox gene lin-39 (Wagmaister et al., 2006b; Guerry et al., 2007) and the Notch pathway ligand gene lag-2 (Zhang and Greenwald, 2011).
Evidence for a LIN-1 activator function is more limited, but unphosphorylated LIN-1 appears to directly activate the Rho kinase gene let-502 during vulva development (Farooqui et al., 2012). Ras-dependent upregulation of lag-2 appears to occur solely through relief of LIN-1-dependent repression (Zhang and Greenwald, 2011).
The Mediator complex: Two widely important positive factors that act in conjunction with LIN-1/Ets are the Mediator subunit SUR-2/Med23 and its partner LIN-25 (Singh and Han, 1995; Tuck and Greenwald, 1995; Nilsson et al., 1998) (Figure 1B). The conserved Mediator complex links certain sequence-specific DNA binding proteins to the general RNA Polymerase II transcriptional machinery (Borggrefe and Yue, 2011). SUR-2 and LIN-25 function together downstream or parallel to MPK-1, and they have very specific Ras-related phenotypes including a partial Vul phenotype and low penetrance defects in excretory duct and P12 development (Singh and Han, 1995; Tuck and Greenwald, 1995; Nilsson et al., 1998; Nilsson et al., 2000). Mammalian Sur2 binds to ERK-phosphorylated Elk1 to recruit Mediator and allow for transcriptional activation of target genes (Boyer et al., 1999; Stevens et al., 2002). The genetics of C. elegans sur-2 and lin-1/Ets are compatible with such a model, though physical interactions between the worm proteins have not been demonstrated and no directly activated targets of phosphorylated LIN-1 or SUR-2 have been identified to date. Analyses of the Ras-responsive lag-2 promoter suggest that, in addition to (or instead of) acting with LIN-1, SUR-2 must function with other unidentified transcription factors (Zhang and Greenwald, 2011).
Another component of the Mediator complex, DPY-22/Med12, functions antagonistically to LET-23/EGFR-Ras-ERK during vulval development, though the precise point at which it acts has not been determined (Moghal and Sternberg, 2003).
EOR-1 and EOR-2: Two other widely important positive factors downstream of MPK-1 are the BTB/Zinc finger protein EOR-1 and its binding partner EOR-2 (Howard and Sundaram, 2002; Howell et al., 2010) (Figure 1B; Table 2). EOR-1 contains two “D-domain” type ERK docking sites and is an excellent substrate of ERK in vitro, and mutation of one of these docking sites or of several potential phosphoacceptors reduces EOR-1 function in vivo (Howell et al., 2010). EOR-1 is related to known transcriptional activators and repressors, including Drosophila GAGA factor and the mammalian oncoproteins PLZF and BCL-6 (Collins et al., 2001; Barna et al., 2002), but it is not known if it acts primarily as an activator or a repressor (or both) or how phosphorylation affects its activity. eor-1 and eor-2 mutants have incompletely penetrant let-60 ras-like defects in excretory duct and P12 development (Rocheleau et al., 2002) and strong defects in age-dependent ubiquitination (Liu et al., 2011), as well as several phenotypes that are apparently unrelated to Ras signaling (Hoeppner et al., 2004). EOR-1 and EOR-2 act redundantly with SUR-2 and LIN-25, such that eor-1; sur-2 double mutants have multiple strong let-60 ras-like loss-of-function phenotypes (Howard and Sundaram, 2002). EOR-1 and EOR-2 also function redundantly with the cryptic positive function of LIN-1, such that lin-1 eor-1 double mutants also have strong let-60 ras-like loss-of-function phenotypes (Howard and Sundaram, 2002; Abdus-Saboor et al., 2011). Thus, together, LIN-1 and EOR-1 appear to mediate many of the somatic effects of Ras signaling.
Hundreds of potential EOR-1 targets have been identified by the modencode project using ChIP-Seq (Niu et al., 2010), but the biological relevance of these targets is still unclear.
LIN-31: The forkhead transcription factor LIN-31 functions downstream of MPK-1 during vulval development (Miller et al., 1993; Tan et al., 1998) and male spicule development (Seah and Sternberg, 2009). LIN-31 appears to be a tissue-specific MPK-1 substrate since it contains a potential ERK docking site (Swathi Arur and Tim Schedl, pers. comm.) and mutation of putative ERK phosphorylation sites in LIN-31 alters function in transgenic assays (Tan et al., 1998). lin-31 mutants have a mixed Vul and Muv phenotype, suggesting that LIN-31 both promotes and inhibits vulval development (Miller et al., 1993). Unphosphorylated LIN-31 appears to bind LIN-1 and inhibit vulval development, while phosphorylated LIN-31 promotes vulval development (Tan et al., 1998). lin-31 has a relatively restricted expression pattern, and ectopic expression of LIN-31 can cause other tissues to adopt some vulval-like characteristics, supporting the model that tissue-specific availability of ERK substrates is one key determinant of tissue-specific responses (Tan et al., 1998; Tan and Kim, 1999).
Hox genes: A common theme among many LET-23/EGFR-Ras-ERK-dependent cell fates is a requirement for specific Hox genes (Figure 1B). In C. elegans, as in other animals, different Hox genes are expressed along the anterior-posterior axis (Aboobaker and Blaxter, 2003). The Hox genes lin-39, egl-5, mab-5 and ceh-13 are upregulated by Ras signaling and/or are required for Ras-dependent induction of vulval, P12, male hook and male spicule fates, respectively (Clandinin et al., 1997; Eisenmann et al., 1998; Maloof and Kenyon, 1998; Wagmaister et al., 2006a; Seah and Sternberg, 2009; Yu et al., 2010). Furthermore, ectopic expression of some Hox genes can change how cells respond to Ras activation (Clandinin et al., 1997; Maloof and Kenyon, 1998; Yu et al., 2010). It therefore appears that the Hox code of a given cell is one important contextual factor that determines tissue-specific outcomes of Ras signaling in the soma.
Germline MPK-1 substrates: In the germline, MPK-1 utilizes a completely different and much broader set of substrates than those described above (Figure 1D; Table 2). Arur et al., (2009) used known ERK docking site sequences (Sharrocks et al., 2000) to identify candidate MPK-1 substrates and then systematically tested their requirements in the germline by RNAi. These studies identified 30 likely MPK-1 substrates that control specific steps of germline development. In general, multiple substrates influenced each step examined, and depletion of individual substrates caused only mild phenotypes, suggesting a high level of substrate redundancy. Many germline MPK-1 substrates also influenced the spatial pattern of MPK-1 phosphorylation, suggesting that they participate in positive or negative feedback loops.
Germline MPK-1 substrates include many different types of proteins (Table 2). MPK-1 substrates relevant to pachytene progression include several RNA helicases, the kinase CDTL-7 and the Rho GAP PAC-1 (Arur et al., 2009). MPK-1 substrates relevant to pachytene cell membrane organization include the translational regulator NOS-3 (related to Drosophila Nanos) (Arur et al., 2011), the DEAD-box helicase DDX-19 (Arur et al., 2009), and the transcription factor DPL-1/DP (Lin and Reinke, 2008). DDX-19 also affects oocyte differentiation (Arur et al., 2009). One of several MPK-1 substrates relevant to oocyte growth control is the kinase GSK-3 (Arur et al., 2009). A potential substrate relevant to DNA-damage induced germline apoptosis is the p53 ortholog CEP-1 (Rutkowski et al., 2011).
The RTK-Ras-ERK pathway often interacts with other signaling pathways to control downstream responses.
Ras-Wnt cooperation: The LET-23/EGFR-Ras-ERK pathway cooperates with a Wnt pathway (see Wnt signaling) to specify vulval fates (Eisenmann et al., 1998; Gleason et al., 2002), P12 fates (Jiang and Sternberg, 1998), and male hook group (Yu et al., 2009) and spicule (Seah and Sternberg, 2009) fates (Figure 8A). The relative importance of Ras vs. Wnt signaling varies among these different tissues–for example, Ras is more important in the vulva and Wnt is more important in the male hook group–but hyperactivation of the “minor” pathway can often bypass the requirement for the “major” pathway, suggesting that both pathways converge on common targets (Gleason et al., 2002; Yu et al., 2009). Some of these common targets may include Hox genes. Interestingly, the relative importance of Ras vs. Wnt signaling also varies between different nematode species - in Pristionchus pacificus, vulval development depends primarily on Wnt signaling (Tian et al., 2008).
|
Figure 8. Interactions between EGF-Ras-ERK and other signaling pathways. A) EGF-Ras-ERK and Wnt signaling cooperate to promote several cell fates. B) EGF-Ras-ERK and Notch signaling function antagonistically to promote distinct cell fates. C) The alternative EGF-Ras-Ral pathway and Notch cooperate to promote the secondary vulval fate. See text for details and references.
Ras-Notch sequential signaling: The LET-23/EGFR-Ras-ERK pathway acts sequentially with a Notch pathway (see LIN-12/Notch signaling in C. elegans) to promote different cell fates during development of the vulva (see Vulval development), uterus (Chang et al., 1999), male hook (Yu et al., 2009) and excretory system (Abdus-Saboor et al., 2011). In the uterus, LIN-12/Notch signaling induces the fate of a progenitor cell (the π cell) (Newman et al., 1995) whose descendants later require Ras signaling to adopt their final fate (uv1) (Chang et al., 1999). In several cases, signaling by one pathway controls the fate of a cell type that expresses a ligand for the other pathway. For example, Ras signaling induces the primary vulval cell type, which then expresses Notch pathway ligands to induce the secondary cell fate (Simske and Kim, 1995; Chen and Greenwald, 2004). LIN-12/Notch signaling inhibits generation of the anchor cell (Greenwald et al., 1983) and LIN-12 and GLP-1/Notch signaling promotes generation of the excretory canal cell (Lambie and Kimble, 1991; Moskowitz and Rothman, 1996), both of which express LIN-3/EGF to induce other cell fates (Hill and Sternberg, 1992; Abdus-Saboor et al., 2011). In these various cases, sequential signaling allows for the properly patterned generation of multiple cell types that make up a functional organ.
Ras-Notch antagonism: When activated within the same cell, LET-23/EGFR-Ras-ERK signaling and Notch signaling tend to function antagonistically (Figure 8B). In primary vulval cells, LET-23/EGFR-Ras-ERK signaling antagonizes LIN-12/Notch signaling by triggering LIN-12 endocytosis (Shaye and Greenwald, 2002). In secondary vulval cells, LIN-12/Notch signaling antagonizes Ras-ERK signaling by stimulating the transcription of various negative regulators such as lip-1 and lst-1-4 (Berset et al., 2001; Yoo et al., 2004). LIN-12/Notch-dependent inhibition of Ras-ERK signaling may allow the alternative Ras-RalGDS pathway to become active, and Notch and Ras-RalGDS can then cooperate to promote the secondary vulval fate (Zand et al., 2011) (Figure 8C).
Coordination of related developmental events: In addition to the Ras-Notch sequential signaling events described above, there are several additional examples where different RTK- or Ras-dependent pathways act together to coordinate related developmental events. For example, after LET-23/EGFR-Ras-ERK-dependent specification of the 1° vulval cell fate, 1° vulval descendants then express EGL-17/FGF and LIN-3/EGF to coordinate sex muscle positioning and uterine development with vulval development (Burdine et al., 1998; Chang et al., 1999). ERK signaling in oocytes and LET-23-PLCγ-IP3 signaling in somatic gonadal cells act together to coordinate oocyte maturation and ovulation (Kim et al., 2012).
Studies in C. elegans have been instrumental in elucidating the basic framework of the RTK-Ras-ERK pathway and many aspects of its regulation and cross-talk with other pathways. It is now clear that each step of the RTK-Ras-ERK pathway is subject to multiple regulatory mechanisms that facilitate or dampen signaling. Many of these mechanisms appear redundant under standard laboratory conditions, such that loss of a single regulatory gene has few phenotypic consequences, and some vary in importance between tissue types. There is also significant redundancy and tissue-specificity among downstream ERK targets. Although much has been learned, many questions remain about how regulatory mechanisms actually work and how use of different ERK targets ultimately leads to the many different and interesting biological outcomes observed.
Importantly, many of the regulatory mechanisms and targets described thus far in C. elegans are widely conserved in other organisms, including humans (Tables 1, 2). Therefore, these represent possible points of intervention in the treatment of diseases that involve aberrant RTK-Ras-ERK signaling. For example, given that hyperactivation of RTKs or Ras is a leading cause of many cancers (Malumbres and Barbacid, 2002; Yoon and Seger, 2006; Karnoub and Weinberg, 2008; Lemmon and Schlessinger, 2010; Udell et al., 2011), drugs targeting positive regulators of the signaling pathway could be one promising approach for cancer therapy. Indeed, antisense oligonucleotides targeting KSR1 have been effective in treating mouse models of pancreatic cancer, effectively suppressing Ras-dependent tumorigenesis with minimal toxicity to wild-type cells (Xing et al., 2003). There are still many challenges to developing clinically-effective drugs, but studies in C. elegans have contributed to a long list of potentially useful targets.
I thank Swathi Arur, Chris Rocheleau, Tim Schedl, and Nathaniel Szewczyk for helpful comments and suggestions on the manuscript. My laboratory's work on RTK-Ras-ERK signaling has been supported by National Institutes of Health grants GM58540 and DK077182.
Abdus-Saboor, I., Mancuso, V.P., Murray, J.I., Palozola, K., Norris, C., Hall, D.H., Howell, K., Huang, K., and Sundaram, M.V. (2011). Notch and Ras promote sequential steps of excretory tube development in C. elegans. Development 138, 3545-3555. Abstract Article
Aboobaker, A., and Blaxter, M. (2003). Hox gene evolution in nematodes: novelty conserved. Curr. Opin. Genet. Dev. 13, 593-598. Abstract Article
Ahearn, I.M., Haigis, K., Bar-Sagi, D., and Philips, M.R. (2012). Regulating the regulator: post-translational modification of RAS. Nat. Rev. Mol. Cell. Biol. 13, 39-51. Abstract Article
Andersen, E.C., and Horvitz, H.R. (2007). Two C. elegans histone methyltransferases repress lin-3 EGF transcription to inhibit vulval development. Development 134, 2991-2999. Abstract Article
Aroian, R.V., Koga, M., Mendel, J.E., Ohshima, Y., and Sternberg, P.W. (1990). The let-23 gene necessary for Caenorhabditis elegans vulval induction encodes a tyrosine kinase of the EGF receptor subfamily [see comments]. Nature 348, 693-699. Abstract Article
Arur, S., Ohmachi, M., Berkseth, M., Nayak, S., Hansen, D., Zarkower, D., and Schedl, T. (2011). MPK-1 ERK controls membrane organization in C. elegans oogenesis via a sex-determination module. Dev. Cell 20, 677-688. Abstract Article
Arur, S., Ohmachi, M., Nayak, S., Hayes, M., Miranda, A., Hay, A., Golden, A., and Schedl, T. (2009). Multiple ERK substrates execute single biological processes in Caenorhabditis elegans germ-line development. Proc. Natl. Acad. Sci. U. S. A. 106, 4776-4781. Abstract Article
Barna, M., Merghoub, T., Costoya, J.A., Ruggero, D., Branford, M., Bergia, A., Samori, B., and Pandolfi, P.P. (2002). Plzf mediates transcriptional repression of HoxD gene expression through chromatin remodeling. Dev. Cell 3, 499-510. Abstract Article
Barolo, S., and Posakony, J.W. (2002). Three habits of highly effective signaling pathways: principles of transcriptional control by developmental cell signaling. Genes Dev. 16, 1167-1181. Abstract Article
Battu, G., Hoier, E.F., and Hajnal, A. (2003). The C. elegans G-protein-coupled receptor SRA-13 inhibits RAS/MAPK signalling during olfaction and vulval development. Development 130, 2567-2577. Abstract Article
Beitel, G.J., Clark, S.G., and Horvitz, H.R. (1990). Caenorhabditis elegans ras gene let-60 acts as a switch in the pathway of vulval induction. Nature 6, 503-509. Abstract Article
Beitel, G.J., Tuck, S., Greenwald, I., and Horvitz, H.R. (1995). The Caenorhabditis elegans gene lin-1 encodes an ETS-domain protein and defines a branch in the vulval induction pathway. Genes Dev. 9, 3149-3162. Abstract Article
Berset, T., Hoier, E.F., Battu, G., Canevascini, S., and Hajnal, A. (2001). Notch inhibition of RAS signaling through MAP kinase phosphatase LIP-1 during C. elegans vulval development. Science 291, 1055-1058. Abstract Article
Berset, T.A., Frohli Hoier, E., and Hajnal, A. (2005). The C. elegans homolog of the tumor suppressor Dep-1/Scc1 inhibits EGFR signaling to regulate binary fate decisions. Genes Dev. 19, 1328-1340. Abstract Article
Birnbaum, D., Popovici, C., and Roubin, R. (2005). A pair as a minimum: the two fibroblast growth factors of the nematode Caenorhabditis elegans. Dev. Dyn. 232, 247-255. Abstract Article
Blobel, C.P., Carpenter, G., and Freeman, M. (2009). The role of protease activity in ErbB biology. Exp. Cell Res. 315, 671-682. Abstract Article
Borggrefe, T., and Yue, X. (2011). Interactions between subunits of the Mediator complex with gene-specific transcription factors. Semin. Cell Dev. Biol. 22, 759-768. Abstract Article
Borland, C.Z., Schutzman, J.L., and Stern, M.J. (2001). Fibroblast growth factor signaling in Caenorhabditis elegans. Bioessays 23, 1120-1130. Abstract Article
Boyer, T.G., Martin, M.E., Lees, E., Ricciardi, R.P., and Berk, A.J. (1999). Mammalian Srb/Mediator complex is targeted by adenovirus E1A protein [see comments]. Nature 399, 276-279. Abstract Article
Branda, C.S., and Stern, M.J. (2000). Mechanisms controlling sex myoblast migration in Caenorhabditis elegans hermaphrodites. Dev. Biol. 226, 137-151. Abstract Article
Bruinsma, J.J., Jirakulaporn, T., Muslin, A.J., and Kornfeld, K. (2002). Zinc ions and cation diffusion facilitator proteins regulate Ras-mediated signaling. Dev. Cell 2, 567-578. Abstract Article
Bui, Y.K., and Sternberg, P.W. (2002). Caenorhabditis elegans inositol 5-phosphatase homolog negatively regulates inositol 1,4,5-triphosphate signaling in ovulation. Mol. Biol. Cell 13, 1641-1651. Abstract Article
Bulow, H.E., Boulin, T., and Hobert, O. (2004). Differential functions of the C. elegans FGF receptor in axon outgrowth and maintenance of axon position. Neuron 42, 367-374. Abstract Article
Bulow, H.E., and Hobert, O. (2006). The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 22, 375-407. Abstract Article
Burdine, R.D., Branda, C.S., and Stern, M.J. (1998). EGL-17(FGF) expression coordinates the attraction of the migrating sex myoblasts with vulval induction in C. elegans. Development 125, 1083-1093. Abstract
Burdine, R.D., Chen, E.B., Kwok, S.F., and Stern, M.J. (1997). egl-17 encodes an invertebrate fibroblast growth factor family member required specifically for sex myoblast migration in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 94, 2433-2437. Abstract Article
Ceol, C.J., and Horvitz, H.R. (2001). dpl-1 DP and efl-1 E2F act with lin-35 Rb to antagonize Ras signaling in C. elegans vulval development. Mol. Cell 7, 461-473. Abstract Article
Ceol, C.J., and Horvitz, H.R. (2004). A new class of C. elegans SynMuv genes implicates a Tip60/NuA4-like HAT complex as a negative regulator of Ras signaling. Dev. Cell 6, 563-576. Abstract Article
Chamberlin, H.M., and Sternberg, P.W. (1994). The lin-3/let-23 pathway mediates inductive signalling during male spicule development in Caenorhabditis elegans. Development 120, 2713-2721. Abstract
Chang, C., Hopper, N.A., and Sternberg, P.W. (2000). Caenorhabditis elegans SOS-1 is necessary for multiple RAS-mediated developmental signals. EMBO J. 19, 3283-3294. Abstract Article
Chang, C., Newman, A.P., and Sternberg, P.W. (1999). Reciprocal EGF signaling back to the uterus from the induced C. elegans vulva coordinates morphogenesis of epithelia. Curr. Biol. 9, 237-246. Abstract Article
Chang, C., and Sternberg, P.W. (1999). C. elegans vulval development as a model system to study the cancer biology of EGFR signaling. Cancer Metastasis Rev. 18, 203-213. Abstract Article
Chateau, M.T., Araiz, C., Descamps, S., and Galas, S. (2010). Klotho interferes with a novel FGF-signalling pathway and insulin/Igf-like signalling to improve longevity and stress resistance in Caenorhabditis elegans. Aging 2, 567-581. Abstract
Chen, L., Fu, Y., Ren, M., Xiao, B., and Rubin, C.S. (2011). A RasGRP, C. elegans RGEF-1b, couples external stimuli to behavior by activating LET-60 (Ras) in sensory neurons. Neuron 70, 51-65. Abstract Article
Chen, L., Ong, B., and Bennett, V. (2001). LAD-1, the Caenorhabditis elegans L1CAM homologue, participates in embryonic and gonadal morphogenesis and is a substrate for fibroblast growth factor receptor pathway-dependent phosphotyrosine-based signaling. J. Cell Biol. 154, 841-855. Abstract Article
Chen, N., and Greenwald, I. (2004). The lateral signal for LIN-12/Notch in C. elegans vulval development comprises redundant secreted and transmembrane DSL proteins. Dev. Cell 6, 183-192. Abstract Article
Chong, H., Lee, J., and Guan, K.-L. (2001). Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 20, 3716-3727. Abstract Article
Church, D., Guan, K.L., and Lambie, E.J. (1995). Three genes of the MAP kinase cascade, mek-2, mpk-1/sur-1 and let-60 ras, are required for meiotic cell cycle progression in Caenorhabditis elegans. Development 121, 2525-2535. Abstract
Clandinin, T.R., DeModena, J.A., and Sternberg, P.W. (1998). Inositol trisphosphate mediates a RAS-independent response to LET-23 receptor tyrosine kinase activation in C. elegans. Cell 92, 523-533. Abstract Article
Clandinin, T.R., Katz, W.S., and Sternberg, P.W. (1997). Caenorhabditis elegans HOM-C genes regulate the response of vulval precursor cells to inductive signal. Dev. Biol. 182, 150-161. Abstract Article
Clark, S.G., Stern, M.J., and Horvitz, H.R. (1992). C. elegans cell-signaling gene sem-5 encodes a protein with SH2 and SH3 domains. Nature 356, 340-344. Abstract Article
Collins, T., Stone, J.R., and Williams, A.J. (2001). All in the family: the BTB/POZ, KRAB and SCAN domains. Mol. Cell. Biol. 21, 3609-3615. Abstract Article
Couteau, F., Guerry, F., Muller, F., and Palladino, F. (2002). A heterochromatin protein 1 homologue in Caenorhabditis elegans acts in germline and vulval development. EMBO Rep. 3, 235-241. Abstract Article
Cui, M., Chen, J., Myers, T.R., Hwang, B.J., Sternberg, P.W., Greenwald, I., and Han, M. (2006). SynMuv genes redundantly inhibit lin-3/EGF expression to prevent inappropriate vulval induction in C. elegans. Dev. Cell 10, 667-672. Abstract Article
Cui, M., and Han, M. (2003). Cis regulatory requirements for vulval cell-specific expression of the Caenorhabditis elegans fibroblast growth factor gene egl-17. Dev. Biol. 257, 104-116. Abstract Article
Davison, E.M., Harrison, M.M., Walhout, A.J., Vidal, M., and Horvitz, H.R. (2005). lin-8, which antagonizes Caenorhabditis elegans Ras-mediated vulval induction, encodes a novel nuclear protein that interacts with the LIN-35 Rb protein. Genetics 171, 1017-1031. Abstract Article
Davison, E.M., Saffer, A.M., Huang, L.S., DeModena, J., Sternberg, P.W., and Horvitz, H.R. (2011). The LIN-15A and LIN-56 transcriptional regulators interact to negatively regulate EGF/Ras signaling in Caenorhabditis elegans vulval cell-fate determination. Genetics 187, 803-815. Abstract Article
Dent, J.A., and Han, M. (1998). Post-embryonic expression pattern of C. elegans let-60 ras reporter constructs. Mech. Dev. 72, 179-182. Abstract Article
DeVore, D.L., Horvitz, H.R., and Stern, M.J. (1995). An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell 83, 611-620. Abstract Article
Dixon, S.J., Alexander, M., Fernandes, R., Ricker, N., and Roy, P.J. (2006). FGF negatively regulates muscle membrane extension in Caenorhabditis elegans. Development 133, 1263-1275. Abstract Article
Dutt, A., Canevascini, S., Froehli-Hoier, E., and Hajnal, A. (2004). EGF signal propagation during C. elegans vulval development mediated by ROM-1 Rhomboid. PLoS Biol. 2, e334. Abstract Article
Eisenmann, D. M., Wnt signaling (June 25, 2005), WormBook, ed. The C. elegans Research Community, WormBook. Abstract Article
Eisenmann, D.M., Maloof, J.N., Simske, J.S., Kenyon, C., and Kim, S.K. (1998). The β-catenin homolog BAR-1 and LET-60 Ras coordinately regulate the Hox gene lin-39 during Caenorhabditis elegans vulval development. Development 125, 3667-3680. Abstract
Fay, D.S., and Yochem, J. (2007). The SynMuv genes of Caenorhabditis elegans in vulval development and beyond. Dev. Biol. 306, 1-9. Abstract Article
Ferguson, E.L., and Horvitz, H.R. (1985). Identification and characterization of 22 genes that affect the vulval cell lineages of the nematode Caenorhabditis elegans. Genetics 110, 17-72. Abstract
Ferguson, E.L., and Horvitz, H.R. (1989). The multivulva phenotype of certain Caenorhabditis elegans mutants results from defects in two functionally redundant pathways. Genetics 123, 109-121. Abstract
Farooqui, S., Pellegrino, M.W., Rimann, I., Morf, M.K., Müller, L., Fröhli, E., and Hajnal, A. (2012). Coordinated lumen contraction and expansion during vulval tube morphogenesis in Caenorhabditis elegans. Dev. Cell. 23, 494-506. Abstract Article
Fixsen, W., Sternberg, P., Ellis, H., and Horvitz, R. (1985). Genes that affect cell fates during the development of Caenorhabditis elegans. Cold Spring Harb. Symp. Quant. Biol. 50, 99-104. Abstract Article
Flores, G.V., Duan, H., Yan, H., Nagaraj, R., Fu, W., Zou, Y., Noll, M., and Banerjee, U. (2000). Combinatorial signaling in the specification of unique cell fates. Cell 103, 75-85. Abstract Article
Gleason, J.E., Korswagen, H.C., and Eisenmann, D.M. (2002). Activation of Wnt signaling bypasses the requirement for RTK/Ras signaling during C. elegans vulval induction. Genes Dev. 16, 1281-1290. Abstract Article
Goldstein, J.L., Glossip, D., Nayak, S., and Kornfeld, K. (2006). The CRAL/TRIO and GOLD domain protein CGR-1 promotes induction of vulval cell fates in Caenorhabditis elegans and interacts genetically with the Ras signaling pathway. Genetics 172, 929-942. Abstract Article
Goodman, S.J., Branda, C.S., Robinson, M.K., Burdine, R.D., and Stern, M.J. (2003). Alternative splicing affecting a novel domain in the C. elegans EGL-15 FGF receptor confers functional specificity. Development 130, 3757-3766. Abstract Article
Gottschalk, A., Almedom, R.B., Schedletzky, T., Anderson, S.D., Yates, J.R., 3rd, and Schafer, W.R. (2005). Identification and characterization of novel nicotinic receptor-associated proteins in Caenorhabditis elegans. EMBO J. 24, 2566-2578. Abstract Article
Govindan, J.A., Cheng, H., Harris, J.E., and Greenstein, D. (2006). Gαo/i and Gαs signaling function in parallel with the MSP/Eph receptor to control meiotic diapause in C. elegans. Curr. Biol. 16, 1257-1268. Abstract Article
Govindan, J.A., Nadarajan, S., Kim, S., Starich, T.A., and Greenstein, D. (2009). Somatic cAMP signaling regulates MSP-dependent oocyte growth and meiotic maturation in C. elegans. Development 136, 2211-2221. Abstract Article
Green, J.L., Inoue, T., and Sternberg, P.W. (2007). The C. elegans ROR receptor tyrosine kinase, CAM-1, non-autonomously inhibits the Wnt pathway. Development 134, 4053-4062. Abstract Article
Green, J.L., Inoue, T., and Sternberg, P.W. (2008). Opposing Wnt pathways orient cell polarity during organogenesis. Cell 134, 646-656. Abstract Article
Greenwald, I. LIN-12/Notch signaling in C. elegans (August 4, 2005), WormBook, ed. The C. elegans Research Community, WormBook. Abstract Article
Greenwald, I.S., Sternberg, P.W., and Horvitz, H.R. (1983). The lin-12 locus specifies cell fates in Caenorhabditis elegans. Cell 34, 435-444. Abstract Article
Gu, T., Orita, S., and Han, M. (1998). Caenorhabditis elegans SUR-5, a novel but conserved protein, negatively regulates LET-60 Ras activity during vulval induction. Mol. Cell. Biol. 18, 4556-4564. Abstract
Guerry, F., Marti, C.-O., Zhang, Y., Moroni, P.S., Jaquiery, E., and Muller, F. (2007). The Mi-2 nucleosome-remodeling protein LET-418 is targeted via LIN-1/ETS to the promoter of lin-39/Hox during vulval development in C. elegans. Dev. Biol. 306, 469-479. Abstract Article
Gumienny, T.L., Lambie, E., Hartwieg, E., Horvitz, H.R., and Hengartner, M.O. (1999). Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 126, 1011-1022. Abstract
Gutch, M.J., Flint, A.J., Keller, J., Tonks, N.K., and Hengartner, M.O. (1998). The Caenorhabditis elegans SH2 domain-containing protein tyrosine phosphatase PTP-2 participates in signal transduction during oogenesis and vulval development. Genes Dev. 12, 571-585. Abstract Article
Hajnal, A., and Berset, T. (2002). The C.elegans MAPK phosphatase LIP-1 is required for the G(2)/M meiotic arrest of developing oocytes. EMBO J. 21, 4317-4326. Abstract Article
Hajnal, A., Whitfield, C.W., and Kim, S.K. (1997). Inhibition of Caenorhabditis elegans vulval induction by gap-1 and by let-23 receptor tyrosine kinase. Genes Dev. 11, 2715-2728. Abstract Article
Halfon, M.S., Carmena, A., Gisselbrecht, S., Sackerson, C.M., Jimenez, F., Baylies, M.K., and Michelson, A.M. (2000). Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell 103, 63-74. Abstract Article
Han, M., Aroian, R.V., and Sternberg, P.W. (1990). The let-60 locus controls the switch between vulval and nonvulval cell fates in Caenorhabditis elegans. Genetics 126, 899-913. Abstract
Han, M., Golden, A., Han, Y., and Sternberg, P.W. (1993). C. elegans lin-45 raf gene participates in let-60 ras-stimulated vulval differentiation. Nature 363, 133-140. Abstract Article
Han, M., and Sternberg, P.W. (1990). let-60, a gene that specifies cell fates during C. elegans vulval induction, encodes a ras protein. Cell 63, 921-931. Abstract Article
Han, M., and Sternberg, P.W. (1991). Analysis of dominant-negative mutations of the Caenorhabditis elegans let-60 ras gene. Genes Dev. 5, 2188-2198. Abstract Article
Han, S.M., Cottee, P.A., and Miller, M.A. (2010). Sperm and oocyte communication mechanisms controlling C. elegans fertility. Dev. Dyn. 239, 1265-1281. Abstract
Hara, M., and Han, M. (1995). Ras farnesyltransferase inhibitors suppress the phenotype resulting from an activated ras mutation in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A. 92, 3333-3337. Abstract Article
Hayashizaki, S., Iino, Y., and Yamamoto, M. (1998). Characterization of the C. elegans gap-2 gene encoding a novel Ras-GTPase activating protein and its possible role in larval development. Genes Cells 3, 189-202. Abstract Article
Hill, R.J., and Sternberg, P.W. (1992). The gene lin-3 encodes an inductive signal for vulval development in C. elegans [see comments]. Nature 358, 470-476. Abstract Article
Hirotsu, T., Saeki, S., Yamamoto, M., and Iino, Y. (2000). The Ras-MAPK pathway is important for olfaction in Caenorhabditis elegans. Nature 404, 289-293. Abstract Article
Hoeppner, D.J., Spector, M.S., Ratliff, T.M., Kinchen, J.M., Granat, S., Lin, S.C., Bhusri, S.S., Conradt, B., Herman, M.A., and Hengartner, M.O. (2004). eor-1 and eor-2 are required for cell-specific apoptotic death in C. elegans. Dev. Biol. 274, 125-138. Abstract Article
Hoffmann, M., Segbert, C., Helbig, G., and Bossinger, O. (2010). Intestinal tube formation in Caenorhabditis elegans requires vang-1 and egl-15 signaling. Dev. Biol. 339, 268-279. Abstract Article
Hopper, N.A. (2006). The adaptor protein soc-1/Gab1 modifies growth factor receptor output in Caenorhabditis elegans. Genetics 173, 163-175. Abstract Article
Hopper, N.A., Lee, J., and Sternberg, P.W. (2000). ARK-1 inhibits EGFR signaling in C. elegans. Mol. Cell 6, 65-75. Abstract Article
Hoskins, R., Hajnal, A.F., Harp, S.A., and Kim, S.K. (1996). The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development 122, 97-111. Abstract
Howard, R.M., and Sundaram, M.V. (2002). C. elegans EOR-1/PLZF and EOR-2 positively regulate Ras and Wnt signaling and function redundantly with LIN-25 and the SUR-2 Mediator complex. Genes Dev. 16, 1815-1827. Abstract Article
Howell, K., Arur, S., Schedl, T., and Sundaram, M.V. (2010). EOR-2 is an obligate binding partner of the BTB-zinc finger protein EOR-1 in Caenorhabditis elegans. Genetics 184, 899-913. Abstract Article
Hsu, V., Zobel, C.L., Lambie, E.J., Schedl, T., and Kornfeld, K. (2002). Caenorhabditis elegans lin-45 raf is essential for larval viability, fertility and the induction of vulval cell fates. Genetics 160, 481-492. Abstract
Hu, P.J. Dauer (August 08, 2007), WormBook, ed. The C. elegans Research Community, WormBook. Abstract Article
Huang, L.S., Tzou, P., and Sternberg, P.W. (1994). The lin-15 locus encodes two negative regulators of Caenorhabditis elegans vulval development. Mol. Biol. Cell 5, 395-411. Abstract
Huang, P., and Stern, M.J. (2004). FGF signaling functions in the hypodermis to regulate fluid balance in C. elegans. Development 131, 2595-2604. Abstract Article
Huang, P., and Stern, M.J. (2005). FGF signaling in flies and worms: more and more relevant to vertebrate biology. Cytokine Growth Factor Rev. 16, 151-158. Abstract Article
Hwang, B.J., and Sternberg, P.W. (2004). A cell-specific enhancer that specifies lin-3 expression in the C. elegans anchor cell for vulval development. Development 131, 143-151. Abstract Article
Inoue, T., Oz, H.S., Wiland, D., Gharib, S., Deshpande, R., Hill, R.J., Katz, W.S., and Sternberg, P.W. (2004). C. elegans LIN-18 is a Ryk ortholog and functions in parallel to LIN-17/Frizzled in Wnt signaling. Cell 118, 795-806. Abstract Article
Iwasa, H., Yu, S., Xue, J., and Driscoll, M. (2010). Novel EGF pathway regulators modulate C. elegans healthspan and lifespan via EGF receptor, PLC-γ, and IP3R activation. Aging Cell 9, 490-505. Abstract
Jacobs, D., Beitel, G.J., Clark, S.G., Horvitz, H.R., and Kornfeld, K. (1998). Gain-of-function mutations in the Caenorhabditis elegans lin-1 ETS gene identify a C-terminal regulatory domain phosphorylated by ERK MAP kinase. Genetics 149, 1809-1822. Abstract
Jacobs, D., Glossip, D., Xing, H., Muslin, A.J., and Kornfeld, K. (1999). Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 13, 163-175. Abstract Article
Jakubowski, J., and Kornfeld, K. (1999). A local, high-density, single-nucleotide polymorphism map used to clone Caenorhabditis elegans cdf-1. Genetics 153, 743-752. Abstract
Jiang, L.I., and Sternberg, P.W. (1998). Interactions of EGF, Wnt and HOM-C genes specify the P12 neuroectoblast fate in C. elegans. Development 125, 2337-2347. Abstract
Johnson, K.G., and Kornfeld, K. (2010). The CRAL/TRIO and GOLD domain protein TAP-1 regulates RAF-1 activation. Dev. Biol. 341, 464-471. Abstract Article
Johnson, S.M., Grosshans, H., Shingara, J., Byrom, M., Jarvis, R., Cheng, A., Labourier, E., Reinert, K.L., Brown, D., and Slack, F.J. (2005). RAS is regulated by the let-7 microRNA family. Cell 120, 635-647. Abstract Article
Jongeward, G.D., Clandinin, T.R., and Sternberg, P.W. (1995). sli-1, a negative regulator of let-23-mediated signaling in C. elegans. Genetics 139, 1553-1566. Abstract
Kaech, S.M., Whitfield, C.W., and Kim, S. (1998). The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell 94, 761-771. Abstract Article
Kamikura, D.M., and Cooper, J.A. (2003). Lipoprotein receptors and a disabled family cytoplasmic adaptor protein regulate EGL-17/FGF export in C. elegans. Genes Dev. 17, 2798-2811. Abstract Article
Kao, G., Tuck, S., Baillie, D., and Sundaram, M.V. (2004). C. elegans SUR-6/PR55 cooperates with LET-92/protein phosphatase 2A and promotes Raf activity independently of inhibitory Akt phosphorylation sites. Development 131, 755-765. Abstract Article
Karnoub, A.E., and Weinberg, R.A. (2008). Ras oncogenes: split personalities. Nat. Rev. Mol. Cell. Biol. 9, 517-531. Abstract Article
Katz, W.S., Hill, R.J., Clandinin, T.R., and Sternberg, P.W. (1995). Different levels of the C. elegans growth factor LIN-3 promote distinct vulval precursor fates. Cell 82, 297-307. Abstract Article
Kim, S., Spike, C., and Greenstein, D. (2012). Control of oocyte growth and meiotic maturation in C. elegans.. Adv. Exp. Med. Biol. 757, 277-320. Abstract Article
Klein, D.E., Nappi, V.M., Reeves, G.T., Shvartsman, S.Y., and Lemmon, M.A. (2004). Argos inhibits epidermal growth factor receptor signalling by ligand sequestration. Nature 430, 1040-1044. Abstract Article
Koga, M., and Ohshima, Y. (1995). Mosaic analysis of the let-23 gene function in vulval induction of Caenorhabditis elegans. Development 121, 2655-2666. Abstract
Kokel, M., Borland, C.Z., DeLong, L., Horvitz, H.R., and Stern, M.J. (1998). clr-1 encodes a receptor tyrosine phosphatase that negatively regulates an FGF receptor signaling pathway in Caenorhabditis elegans. Genes Dev. 12, 1425-1437. Abstract Article
Kornfeld, K., Guan, K.L., and Horvitz, H.R. (1995a). The Caenorhabditis elegans gene mek-2 is required for vulval induction and encodes a protein similar to the protein kinase MEK. Genes Dev. 9, 756-768. Abstract Article
Kornfeld, K., Hom, D.B., and Horvitz, H.R. (1995b). The ksr-1 gene encodes a novel protein kinase involved in Ras-mediated signaling in Caenorhabditis elegans. Cell 83, 903-913. Abstract Article
Kritikou, E.A., Milstein, S., Vidalain, P.O., Lettre, G., Bogan, E., Doukoumetzidis, K., Gray, P., Chappell, T.G., Vidal, M., and Hengartner, M.O. (2006). C. elegans GLA-3 is a novel component of the MAP kinase MPK-1 signaling pathway required for germ cell survival. Genes Dev. 20, 2279-2292. Abstract Article
Lackner, M.R., and Kim, S.K. (1998). Genetic analysis of the Caenorhabditis elegans MAP kinase gene mpk-1. Genetics 150, 103-117. Abstract
Lackner, M.R., Kornfeld, K., Miller, L.M., Horvitz, H.R., and Kim, S.K. (1994). A MAP kinase homolog, mpk-1, is involved in ras-mediated induction of vulval cell fates in Caenorhabditis elegans. Genes Dev. 8, 160-173. Abstract Article
Lambie, E.J., and Kimble, J. (1991). Two homologous regulatory genes, lin-12 and glp-1, have overlapping functions. Development 112, 231-240. Abstract
Lamitina, T., Huang, C.G., and Strange, K. (2006). Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc. Natl. Acad. Sci. U. S. A. 103, 12173-12178. Abstract Article
Lanigan, T.M., Liu, A., Huang, Y.Z., Mei, L., Margolis, B., and Guan, K.L. (2003). Human homologue of Drosophila CNK interacts with Ras effector proteins Raf and Rlf. FASEB J. 17, 2048-2060. Abstract Article
Lee, J., Jongeward, G.D., and Sternberg, P.W. (1994). unc-101, a gene required for many aspects of Caenorhabditis elegans development and behavior, encodes a clathrin-associated protein. Genes Dev. 8, 60-73. Abstract Article
Lee, Myon-Hee, Hook, B., Lamont, L.B., Wickens, M., and Kimble, J. (2006). LIP-1 phosphatase controls the extent of germline proliferation in Caenorhabditis elegans. EMBO J. 25, 88-96. Abstract Article
Lee, Myon-Hee, Hook, B., Pan, G., Kershner, A.M., Merritt, C., Seydoux, G., Thomson, J.A., Wickens, M., and Kimble, J. (2007). Conserved regulation of MAP kinase expression by PUF RNA-binding proteins. PLoS Genet. 3, e233. Abstract Article
Lee, Min-Ho, Ohmachi, M., Arur, S., Nayak, S., Francis, R., Church, D., Lambie, E., and Schedl, T. (2007). Multiple functions and dynamic activation of MPK-1 extracellular signal-regulated kinase signaling in Caenorhabditis elegans germline development. Genetics 177, 2039-2062. Abstract Article
Leight, E.R., Glossip, D., and Kornfeld, K. (2005). Sumoylation of LIN-1 promotes transcriptional repression and inhibition of vulval cell fates. Development 132, 1047-1056. Abstract Article
Lemmon, M.A., and Schlessinger, J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117-1134. Abstract Article
Li, W., Han, M., and Guan, K.L. (2000). The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev. 14, 895-900. Abstract
Lin, B., and Reinke, V. (2008). The candidate MAP kinase phosphorylation substrate DPL-1 (DP) promotes expression of the MAP kinase phosphatase LIP-1 in C. elegans germ cells. Dev. Biol. 316, 50-61. Abstract Article
Liu, G., Rogers, J., Murphy, C.T., and Rongo, C. (2011). EGF signalling activates the ubiquitin proteasome system to modulate C. elegans lifespan. EMBO J. 30, 2990-3003. Abstract Article
Lo, T.W., Bennett, D.C., Goodman, S.J., and Stern, M.J. (2010). Caenorhabditis elegans fibroblast growth factor receptor signaling can occur independently of the multi-substrate adaptor FRS2. Genetics 185, 537-547. Abstract Article
Lo, T.W., Branda, C.S., Huang, P., Sasson, I.E., Goodman, S.J., and Stern, M.J. (2008). Different isoforms of the C. elegans FGF receptor are required for attraction and repulsion of the migrating sex myoblasts. Dev. Biol. 318, 268-275. Abstract Article
Maloof, J.N., and Kenyon, C. (1998). The Hox gene lin-39 is required during C. elegans vulval induction to select the outcome of Ras signaling. Development 125, 181-190. Abstract
Malumbres, M., and Barbacid, M. (2002). RAS oncogenes: the first 30 years. Nat. Rev. Cancer 3, 7-13. Abstract
Manning, G. Genomic overview of protein kinases (December 13, 2005), WormBook, ed. The C. elegans Research Community, WormBook. Abstract Article
Matsubara, Y., Kawasaki, I., Urushiyama, S., Yasuda, T., Shirakata, M., Iino, Y., Shibuya, H., and Yamanashi, Y. (2007). The adaptor-like protein ROG-1 is required for activation of the Ras-MAP kinase pathway and meiotic cell cycle progression in Caenorhabditis elegans. Genes Cells 12, 407-420. Abstract Article
McCarter, J. (1998). The regulation of oocyte maturation and ovulation in Caenorhabditis elegans. PhD Thesis, Washington University in St. Louis, St Louis MO.
McCarter, J., Bartlett, B., Dang, T., and Schedl, T. (1999). On the control of oocyte meiotic maturation and ovulation in Caenorhabditis elegans. Dev. Biol. 205, 111-128. Abstract Article
McMullan, R., Anderson, A., and Nurrish, S. (2012). Behavioral and immune responses to infection require Gaq-RhoA signaling in C. elegans. PLoS Pathog. 8, e1002530. Abstract Article
Merz, D.C., Alves, G., Kawano, T., Zheng, H., and Culotti, J.G. (2003). UNC-52/perlecan affects gonadal leader cell migrations in C. elegans hermaphrodites through alterations in growth factor signaling. Dev. Biol. 256, 173-186. Abstract Article
Miller, L.M., Gallegos, M.E., Morisseau, B.A., and Kim, S. (1993). lin-31, a Caenorhabditis elegans HNF-3/forkhead transcription factor homolog, specificies three alternative cell fates in vulval development. Genes Dev. 7, 933-947. Abstract Article
Miller, M.A., Nguyen, V.Q., Lee, M.H., Kosinski, M., Schedl, T., Caprioli, R.M., and Greenstein, D. (2001). A sperm cytoskeletal protein that signals oocyte meiotic maturation and ovulation. Science 291, 2144-2147. [see comments]. [erratum appears in Science 2001 292, 639; Science 2001 292, 53]. Abstract
Miller, M.A., Ruest, P.J., Kosinski, M., Hanks, S.K., and Greenstein, D. (2003). An Eph receptor sperm-sensing control mechanism for oocyte meiotic maturation in Caenorhabditis elegans. Genes Dev. 17, 187-200. Abstract Article
Miura, G.I., Buglino, J., Alvarado, D., Lemmon, M.A., Resh, M.D., and Treisman, J.E. (2006). Palmitoylation of the EGFR ligand Spitz by Rasp increases Spitz activity by restricting its diffusion. Dev. Cell 10, 167-176. Abstract Article
Moghal, N., Garcia, L.R., Khan, L.A., Iwasaki, K., and Sternberg, P.W. (2003). Modulation of EGF receptor-mediated vulva development by the heterotrimeric G-protein Gαq and excitable cells in C. elegans. Development 130, 4553-4566. Abstract
Moghal, N., and Sternberg, P.W. (2003). A component of the transcriptional mediator complex inhibits RAS-dependent vulval fate specification in C. elegans. Development 130, 57-69. Abstract Article
Moskowitz, I.P., and Rothman, J.H. (1996). lin-12 and glp-1 are required zygotically for early embryonic cellular interactions and are regulated by maternal GLP-1 signaling in Caenorhabditis elegans. Development 122, 4105-4117. Abstract
Muller, J., Ory, S., Copeland, T., Piwnica-Worms, H., and Morrison, D.K. (2001). C-TAK1 regulates Ras signaling by phosphorylating the MAPK scaffold KSR1. Mol. Cell 8, 983-993. Abstract Article
Myers, T.R., and Greenwald, I. (2005). lin-35 Rb acts in the major hypodermis to oppose Ras-mediated vulval induction in C. elegans. Dev. Cell 8, 117-123. Abstract Article
Nanji, M., Hopper, N.A., and Gems, D. (2005). LET-60 RAS modulates effects of insulin/IGF-1 signaling on development and aging in Caenorhabditis elegans. Aging Cell 4, 235-245. Abstract Article
Nelson, F.K., Albert, P.S., and Riddle, D.L. (1983). Fine structure of the C. elegans secretory-excretory system. J. Ultrastruct. Res. 82, 156-171. Abstract Article
Nelson, F.K., and Riddle, D.L. (1984). Functional study of the C. elegans secretory-excretory system using laser microsurgery. J. Exp. Zool. 231, 45-56. Abstract Article
Newman, A.P., White, J.G., and Sternberg, P.W. (1995). The Caenorhabditis elegans lin-12 gene mediates induction of ventral uterine specialization by the anchor cell. Development 121, 263-271. Abstract
Nicholas, H.R., and Hodgkin, J. (2004). The ERK MAP kinase cascade mediates tail swelling and a protective response to rectal infection in C. elegans. Curr. Biol. 14, 1256-1261. Abstract Article
Nilsson, L., Li, X., Tiensuu, T., Auty, R., Greenwald, I., and Tuck, S. (1998). Caenorhabditis elegans lin-25: cellular focus, protein expression and requirement for sur-2 during induction of vulval fates. Development 125, 4809-4819. Abstract
Nilsson, L., Tiensuu, T., and Tuck, S. (2000). Caenorhabditis elegans lin-25: a study of its role in multiple cell fate specification events involving Ras and the identification and characterization of evolutionarily conserved domains. Genetics 156, 1083-1096. Abstract
Niu, W., Lu, Z.J., Zhong, M., Sarov, M., Murray, J.I., Brdlik, C.M., Janette, J., Chen, C., Alves, P., Preston, E., et al. (2010). Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res. 21, 245-254. Abstract Article
Nykamp, K., Lee, M.H., and Kimble, J. (2008). C. elegans La-related protein, LARP-1, localizes to germline P bodies and attenuates Ras-MAPK signaling during oogenesis. RNA 14, 1378-1389. Abstract Article
Ohmachi, M., Rocheleau, C.E., Church, D., Lambie, E., Schedl, T., and Sundaram, M.V. (2002). C. elegans ksr-1 and ksr-2 have both unique and redundant functions and are required for MPK-1 ERK phosphorylation. Curr. Biol. 12, 427-433. Abstract Article
Okuyama, T., Inoue, H., Ookuma, S., Satoh, T., Kano, K., Honjoh, S., Hisamoto, N., Matsumoto, K., and Nishida, E. (2010). The ERK-MAPK pathway regulates longevity through SKN-1 and insulin-like signaling in Caenorhabditis elegans. J. Biol. Chem. 285, 30274-30281. Abstract Article
Ory, S., Zhou, M., Conrads, T.P., Veenstra, T.D., and Morrison, D.K. (2003). Protein Phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol. 13, 1356-1364. Abstract Article
Page, B.D., Guedes, S., Waring, D., and Priess, J.R. (2001). The C. elegans E2F- and DP-related proteins are required for embryonic asymmetry and negatively regulate Ras/MAPK signaling. Mol. Cell 7, 451-460. Abstract Article
Pizette, S., Rabouille, C., and Cohen, S.M. (2009). Glycosphingolipids control the extracellular gradient of the Drosophila EGFR ligand Gurken. Development 136, 551-561. Abstract Article
Polanska, U.M., Duchesne, L., Harries, J.C., Fernig, D.G., and Kinnunen, T.K. (2009). N-Glycosylation regulates fibroblast growth factor receptor/EGL-15 activity in Caenorhabditis elegans in vivo. J. Biol. Chem. 284, 33030-33039. Abstract Article
Polanska, U.M., Edwards, E., Fernig, D.G., and Kinnunen, T.K. (2011). The cooperation of FGF receptor and Klotho is involved in excretory canal development and regulation of metabolic homeostasis in Caenorhabditis elegans. J. Biol. Chem. 286, 5657-5666. Abstract Article
Popovici, C., Conchonaud, F., Birnbaum, D., and Roubin, R. (2004). Functional phylogeny relates LET-756 to fibroblast growth factor 9. J. Biol. Chem. 279, 40146-40152. Abstract Article
Popovici, C., Fallet, M., Marguet, D., Birnbaum, D., and Roubin, R. (2006). Intracellular trafficking of LET-756, a fibroblast growth factor of C. elegans, is controlled by a balance of export and nuclear signals. Exp. Cell Res. 312, 1484-1495. Abstract Article
Popovici, C., Roubin, R., Coulier, F., Pontarotti, P., and Birnbaum, D. (1999). The family of Caenorhabditis elegans tyrosine kinase receptors: similarities and differences with mammalian receptors. Genome Res. 9, 1026-1039. Abstract Article
Reiner, D.J., Ailion, M., Thomas, J.H., and Meyer, B.J. (2008). C. elegans anaplastic lymphoma kinase ortholog SCD-2 controls dauer formation by modulating TGF-β signaling. Curr. Biol. 18, 1101-1109. Abstract
Rocheleau, C.E., Howard, R.M., Goldman, A.P., Volk, M.L., Girard, L.J., and Sundaram, M.V. (2002). A lin-45 raf enhancer screen identifies eor-1, eor-2 and unusual alleles of Ras pathway genes in Caenorhabditis elegans. Genetics 161, 121-131. Abstract
Rocheleau, C.E., Ronnlund, A., Tuck, S., and Sundaram, M.V. (2005). Caenorhabditis elegans CNK-1 promotes Raf activation but is not essential for Ras/Raf signaling. Proc. Natl. Acad. Sci. U. S. A. 102, 11757-11762. Abstract Article
Roubin, R., Naert, K., Popovici, C., Vatcher, G., Coulier, F., Thierry-Mieg, J., Pontarotti, P., Birnbaum, D., Baillie, D., and Thierry-Mieg, D. (1999). let-756, a C. elegans fgf essential for worm development. Oncogene 18, 6741-6747. Abstract Article
Ruivenkamp, C.A., van Wezel, T., Zanon, C., Stassen, A.P., Vlcek, C., Csikos, T., Klous, A.M., Tripodis, N., Perrakis, A., Boerrigter, L., et al. (2002). Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat. Genet. 31, 295-300. Abstract Article
Rutkowski, R., Dickinson, R., Stewart, G., Craig, A., Schimpl, M., Keyse, S.M., and Gartner, A. (2011). Regulation of Caenorhabditis elegans p53/CEP-1-dependent germ cell apoptosis by Ras/MAPK signaling. PLoS Genet. 7, e1002238. Abstract Article
Saffer, A.M., Kim, D.H., van Oudenaarden, A., and Horvitz, H.R. (2011). The Caenorhabditis elegans synthetic multivulva genes prevent Ras pathway activation by tightly repressing global ectopic expression of lin-3 EGF. PLoS Genet. 7, e1002418. Abstract Article
Sasson, I.E., and Stern, M.J. (2004). FGF and PI3 kinase signaling pathways antagonistically modulate sex muscle differentiation in C. elegans. Development 131, 5381-5392. Abstract Article
Schouest, K.R., Kurasawa, Y., Furuta, T., Hisamoto, N., Matsumoto, K., and Schumacher, J.M. (2009). The germinal center kinase GCK-1 is a negative regulator of MAP kinase activation and apoptosis in the C. elegans germline. PLoS One 4, e7450. Abstract Article
Schutzman, J.L., Borland, C.Z., Newman, J.C., Robinson, M.K., Kokel, M., and Stern, M.J. (2001). The Caenorhabditis elegans EGL-15 signaling pathway implicates a DOS-like multisubstrate adaptor protein in fibroblast growth factor signal transduction. Mol. Cell. Biol. 21, 8104-8116. Abstract Article
Seah, A., and Sternberg, P.W. (2009). The roles of EGF and Wnt signaling during patterning of the C. elegans Bγ/δ Equivalence Group. BMC Dev. Biol. 9, 74. Abstract
Selfors, L.M., Schutzman, J.L., Borland, C.Z., and Stern, M.J. (1998). soc-2 encodes a leucine-rich repeat protein implicated in fibroblast growth factor receptor signaling. Proc. Natl. Acad. Sci. U. S. A. 95, 6903-6908. Abstract Article
Sharrocks, A.D. (2002). Complexities in ETS-domain transcription factor function and regulation: lessons from the TCF (ternary complex factor) subfamily. Biochem. Soc. Trans. 30, 1-9. Abstract
Sharrocks, A.D., Yang, S.H., and Galanis, A. (2000). Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem. Sci. 25, 448-453. Abstract Article
Shaye, D.D., and Greenwald, I. (2002). Endocytosis-mediated downregulation of LIN-12/Notch upon Ras activation in Caenorhabditis elegans. Nature 420, 686-690. Abstract Article
Shen, F., Lin, Q., Gu, Y., Childress, C., and Yang, W. (2007). Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol. Biol. Cell 18, 732-742. Abstract Article
Shibatohge, M., Kariya, K., Liao, Y., Hu, C.D., Watari, Y., Goshima, M., Shima, F., and Kataoka, T. (1998). Identification of PLC210, a Caenorhabditis elegans phospholipase C, as a putative effector of Ras. J. Biol. Chem. 273, 6218-6222. Abstract Article
Sieburth, D.S., Sun, Q., and Han, M. (1998). SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94, 119-130. Abstract Article
Sieburth, D.S., Sundaram, M., Howard, R.M., and Han, M. (1999). A PP2A regulatory subunit positively regulates Ras-mediated signaling during Caenorhabditis elegans vulval induction. Genes Dev. 13, 2562-2569. Abstract Article
Simske, J.S., Kaech, S.M., Harp, S.A., and Kim, S.K. (1996). LET-23 receptor localization by the cell junction protein LIN-7 during C. elegans vulval induction. Cell 85, 195-204. Abstract Article
Simske, J.S., and Kim, S.K. (1995). Sequential signalling during Caenorhabditis elegans vulval induction. Nature 375, 142-146. Abstract Article
Singh, N., and Han, M. (1995). sur-2, a novel gene, functions late in the let-60 ras-mediated signaling pathway during Caenorhabditis elegans vulval induction. Genes Dev. 9, 2251-2265. Abstract Article
Skorobogata, O., and Rocheleau, C.E. (2012). RAB-7 antagonizes LET-23 EGFR signaling during vulva development in Caenorhabditis elegans. PLoS One 7, e36489. Abstract Article
Solari, F., and Ahringer, J. (2000). NURD-complex genes antagonise Ras-induced vulval development in Caenorhabditis elegans. Curr. Biol. 10, 223-226. Abstract Article
Stern, M.J., and Horvitz, H.R. (1991). A normally attractive cell interaction is repulsive in two C. elegans mesodermal cell migration mutants. Development 113, 797-803. Abstract
Sternberg, P.W., Vulval development (June, 25 2005), WormBook, ed. The C. elegans Research Community, WormBook. Abstract Article
Stetak, A., Gutierrez, P., and Hajnal, A. (2008). Tissue-specific functions of the Caenorhabditis elegans p120 Ras GTPase activating protein GAP-3. Dev. Biol. 323, 166-176. Abstract Article
Stetak, A., Hoier, E.F., Croce, A., Cassata, G., Di Fiore, P.P., and Hajnal, A. (2006). Cell fate-specific regulation of EGF receptor trafficking during Caenorhabditis elegans vulval development. EMBO J. 25, 2347-2357. Abstract Article
Stevens, J.L., Cantin, G.T., Wang, G., Shevchenko, A., and Berk, A.J. (2002). Transcription control by E1A and MAP kinase pathway via Sur2 mediator subunit. Science 296, 755-758. Abstract Article
Stewart, S., Sundaram, M., Zhang, Y., Lee, J., Han, M., and Guan, K.L. (1999). Kinase Suppressor of Ras (KSR) forms a multi-protein signaling complex and modulates MEK localization. Mol. Cell. Biol. 19, 5523-5534. Abstract
Stone, C.E., Hall, D.H., and Sundaram, M.V. (2009). Lipocalin signaling controls unicellular tube development in the Caenorhabditis elegans excretory system. Dev. Biol. 329, 201-211. Abstract Article
Sulston, J.E., Albertson, D.G., and Thomson, J.N. (1980). The Caenorhabditis elegans male: postembryonic development of nongonadal structures. Dev. Biol. 78, 542-576. Abstract Article
Sulston, J.E., and Horvitz, H.R. (1977). Post-embryonic cell lineages of the nematode Caenorhabditis elegans. Dev. Biol. 56, 110-156. Abstract Article
Sulston, J.E., Scheirenberg, E., White, J.G., and Thomson, J.N. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 100, 64-119. Abstract Article
Sulston, J.E., and White, J.G. (1980). Regulation and cell autonomy during postembryonic development of Caenorhabditis elegans. Dev. Biol. 78, 577-597. Abstract Article
Sundaram, M., and Han, M. (1995). The C. elegans ksr-1 gene encodes a novel Raf-related kinase involved in Ras-mediated signal transduction. Cell 83, 889-901. Abstract Article
Sundaram, M., Yochem, J., and Han, M. (1996). A Ras-mediated signal transduction pathway is involved in the control of sex myoblast migration in Caenorhabditis elegans. Development 122, 2823-2833. Abstract
Szewczyk, N.J., and Jacobson, L.A. (2003). Activated EGL-15 FGF receptor promotes protein degradation in muscles of Caenorhabditis elegans. EMBO J. 22, 5058-5067. Abstract Article
Szewczyk, N.J., Peterson, B.K., and Jacobson, L.A. (2002). Activation of Ras and the mitogen-activated protein kinase pathway promotes protein degradation in muscle cells of Caenorhabditis elegans. Mol. Cell. Biol. 22, 4181-4188. Abstract Article
Tan, P., and Kim, S.K. (1999). Signaling specificity: the RTK/Ras/MAP kinase pathway in metazoans. Trends Genet. 15, 145-149. Abstract Article
Tan, P.B., Lackner, M.R., and Kim, S.K. (1998). MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell 93, 569-580. Abstract Article
Tarcic, G., Boguslavsky, S.K., Wakim, J., Kiuchi, T., Liu, A., Reinitz, F., Nathanson, D., Takahashi, T., Mischel, P.S., Ng, T., et al. (2009). An unbiased screen identifies DEP-1 tumor suppressor as a phosphatase controlling EGFR endocytosis. Curr. Biol. 19, 1788-1798. Abstract Article
Tian, H., Schlager, B., Xiao, H., and Sommer, R.J. (2008). Wnt signaling induces vulva development in the nematode Pristionchus pacificus. Curr. Biol. 18, 142-146. Abstract Article
Tidyman, W.E., and Rauen, K.A. (2009). The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr. Opin. Genet. Dev. 19, 230-236. Abstract Article
Tiensuu, T., Larsen, M.K., Vernerrson, E., and Tuck, S. (2005). lin-1 has both positive and negative functions in specifying multiple cell fates induced by Ras/MAPK signaling in C. elegans. Dev. Biol. 286, 338-351. Abstract Article
Tornberg, J., Sykiotis, G.P., Keefe, K., Plummer, L., Hoang, X., Hall, J.E., Quinton, R., Seminara, S.B., Hughes, V., Van Vliet, G., et al. (2011). Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. U. S. A. 108, 11524-11529. Abstract Article
Tuck, S., and Greenwald, I. (1995). lin-25, a gene required for vulval induction in Caenorhabditis elegans. Genes Dev. 9, 341-357. Abstract Article
Udell, C.M., Rajakulendran, T., Sicheri, F., and Therrien, M. (2011). Mechanistic principles of RAF kinase signaling. Cell. Mol. Life Sci. 68, 553-565. Abstract Article
Van Buskirk, C., and Sternberg, P.W. (2007). Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegans. Nature Neurosci. 10, 1300-1307. Abstract Article
Wagmaister, J.A., Gleason, J.E., and Eisenmann, D.M. (2006a). Transcriptional upregulation of the C. elegans Hox gene lin-39 during vulval cell fate specification. Mech. Dev. 123, 135-150. Abstract Article
Wagmaister, J.A., Miley, G.R., Morris, C.A., Gleason, J.E., Miller, L.M., Kornfeld, K., and Eisenmann, D.M. (2006b). Identification of cis-regulatory elements from the C. elegans Hox gene lin-39 required for embryonic expression and for regulation by the transcription factors LIN-1, LIN-31 and LIN-39. Dev. Biol. 297, 550-565. Abstract Article
Watari, Y., Kariya, K., Shibatohge, M., Liao, Y., Hu, C.D., Goshima, M., Tamada, M., Kikuchi, A., and Kataoka, T. (1998). Identification of Ce-AF-6, a novel Caenorhabditis elegans protein, as a putative Ras effector. Gene 224, 53-58. Abstract
Whitfield, C.W., Benard, C., Barnes, T., Hekimi, S., and Kim, S.K. (1999). Basolateral localization of the Caenorhabditis elegans epidermal growth factor receptor in epithelial cells by the PDZ protein LIN-10. Mol. Biol. Cell 10, 2087-2100. Abstract
Wu, Y., and Han, M. (1994). Suppression of activated Let-60 ras protein defines a role of Caenorhabditis elegans Sur-1 MAP kinase in vulval differentiation. Genes Dev. 8, 147-159. Abstract Article
Wu, Y., Han, M., and Guan, K.L. (1994). MEK-2, a Caenorhabditis elegans MAP kinase kinase, functions in Ras-mediated vulval induction and other developmental events. Genes Dev. 9, 742-755. Abstract Article
Xing, H., Kornfeld, K., and Muslin, A.J. (1997). The protein kinase KSR interacts with 14-3-3 protein and Raf. Curr. Biol. 7, 294-300. Abstract Article
Xing, H. R., Cordon-Cardo, C., Deng, X., Tong, W., Campodonico, L., Fuks, Z., Kolesnick, R. (2003). Pharmacologic inactivation of kinase suppressor of ras-1 abrogates Ras-mediated pancreatic cancer. Nat. Med. 9, 1267-1268. Abstract Article
Yang, S.H., Jaffray, E., Hay, R.T., and Sharrocks, A.D. (2003). Dynamic interplay of the SUMO and ERK pathways in regulating Elk-1 transcriptional activity. Mol. Cell 12, 63-74. Abstract Article
Yang, Y., Han, S.M., and Miller, M.A. (2010). MSP hormonal control of the oocyte MAP kinase cascade and reactive oxygen species signaling. Dev. Biol. 342, 96-107. Abstract Article
Yao, I., Ohtsuka, T., Kawabe, H., Matsuura, Y., Takai, Y., and Hata, Y. (2000). Association of membrane-associated guanylate kinase-interacting protein-1 with Raf-1. Biochem. Biophys. Res. Commun. 270, 538-542. Abstract Article
Yin, X., Gower, N.J., Baylis, H.A., and Strange, K. (2004). Inositol 1,4,5-triphosphate signaling regulates rhythmic contractile activity of myoepithelial sheath cells in Caenorhabditis elegans. Mol. Biol. Cell 15, 3938-3949. Abstract Article
Yochem, J., Sundaram, M., and Han, M. (1997). Ras is required for a limited number of cell fates and not for general proliferation in Caenorhabditis elegans. Mol. Cell. Biol. 17, 2716-2722. Abstract
Yoder, J.H., Chong, H., Guan, K.L., and Han, M. (2004). Modulation of KSR activity in Caenorhabditis elegans by Zn ions, PAR-1 kinase and PP2A phosphatase. EMBO J. 23, 111-119. Abstract Article
Yoo, A.S., Bais, C., and Greenwald, I. (2004). Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science 303, 663-666. Abstract Article
Yoon, C.H., Lee, J., Jongeward, G.D., and Sternberg, P.W. (1995). Similarity of sli-1, a regulator of vulval development in C. elegans, to the mammalian proto-oncogene c-cbl. Science 269, 1102-1105. Abstract Article
Yoon, S., and Seger, R. (2006). The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21-44. Abstract Article
Yu, H., Seah, A., Herman, M.A., Ferguson, E.L., Horvitz, H.R., and Sternberg, P.W. (2009). Wnt and EGF pathways act together to induce C. elegans male hook development. Dev. Biol. 327, 419-432. Abstract Article
Yu, H., Seah, A., and Sternberg, P.W. (2010). Re-programming of C. elegans male epidermal precursor fates by Wnt, Hox, and LIN-12/Notch activities. Dev. Biol. 345, 1-11. Abstract Article
Yu, W., Fantl, W.J., Harrowe, G., and Williams, L.T. (1998). Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr. Biol. 8, 56-64. Abstract Article
*Edited by: Iva Greenwald. Last revised March 1, 2013, Published July 1, 2013. This chapter should be cited as: Sundaram M.V. Canonical RTK-Ras-ERK signaling and related alternative pathways (July 1, 2013), WormBook, ed. The C. elegans Research Community, WormBook, doi/10.1895/wormbook.1.80.2, http://www.wormbook.org.
Copyright: © 2013 Meera V. Sundaram. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
§To whom correspondence should be addressed. E-mail: sundaram@mail.med.upenn.edu
 All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.
All WormBook content, except where otherwise noted, is licensed under a Creative Commons Attribution License.